Lidstrom: SDS-PAGE: Difference between revisions
(→Ladder) |
|||
| (115 intermediate revisions by 3 users not shown) | |||
| Line 1: | Line 1: | ||
Return: [[Lidstrom:Protocols | Protocols]] | Return: [[Lidstrom:Protocols | Lidstrom Lab Protocols]] | ||
Return: [[SDS-PAGE| official OpenWetWare SDS-PAGE]] | |||
Contact [[User:Janet B. Matsen|Janet Matsen]] with questions, comments, & corrections. | |||
== Intro == | |||
* [http://sdspage.homestead.com/ the very basics] | |||
=='''Gel Prep'''== | =='''Gel Prep'''== | ||
* Clean cover plate and thicker spacer plate 75 mM | * Clean cover plate and thicker spacer plate (75 mM gap) | ||
** Soap and Water | ** Soap and Water: scrub with gloved fingers. Ensure all chunks are gone. | ||
** | ** Rinse with water, then ethanol. Prop up vertically against green clamps for stands so they drain and dry. | ||
* Setup one spacer plate and one cover plate in each gel holder | * Setup one spacer plate and one cover plate in each gel holder | ||
** The cover plate goes on the side of the spacer plate with the spacers in order to create a small gap between the plates | ** The cover plate goes on the side of the spacer plate with the spacers in order to create a small gap between the plates | ||
** Put on the black counter top to pinch closed. Don't put it on the absorbent mat, as this slight unevenness will increase your gel leakage rate. Janet had ~33% of gels leak until she started doing it on the black counter without padding; now she has ~5-10% of poured gels leak. | |||
* Put the gel holder into the casting stand | * Put the gel holder into the casting stand | ||
* '''Make a fresh 10% (w/vol) APS solution'''. Don't use an "old" solution; the gel won't polymerize. 10% w/v = 0.1 g/mL. | * '''Make a fresh 10% (w/vol) APS solution'''. Don't use an "old" solution; the gel won't polymerize. 10% w/v = 0.1 g/mL. | ||
** 1 month old seems to be safe, but up to 3 months may be ok. | |||
=== '''Pour the Resolving Gel'''=== | === '''Pour the Resolving Gel'''=== | ||
| Line 16: | Line 23: | ||
*[http://mach7.bluehill.com/proteinc/tutorial/sdspage.html Gel Mix Recipe] | *[http://mach7.bluehill.com/proteinc/tutorial/sdspage.html Gel Mix Recipe] | ||
** Don't add APS/TEMED until ready to pour | ** Don't add APS/TEMED until ready to pour | ||
** '''The APS solution should be | ** '''The APS solution should be < 1 month old.''' Up to 2 months might be ok, but we don't have great data for this. | ||
* Use pipette to put gel mix into the gap between the plates | *** A 3 month old solution failed to cause polymerization & the liquid spilled out from between the plates when released from the stand. -[[User:Janet B. Matsen|JM 10/2012]] | ||
*** The BioRad protocols insist you should make it fresh every time, but we never do if we have one made within the last 4 weeks | |||
* Use pipette to put gel mix into the gap between the plates. Fill to about the bottom of the green line that is below the short plate. | |||
* '''leave room for comb + 1 cm of resolving gel''' (citation: [http://webcache.googleusercontent.com/search?q=cache:SWfzFRUgWxUJ:https://www.lablife.org/g?a%3Divy_file%26fh%3DYXvc5936lyS6GTqz6vTO1JNeQgY-%26v%3D1%26id%3Dg2441.CgrzyhkmAUhlRXEHPWLRnldxQuk-%26f%3Dattachments+&cd=7&hl=en&ct=clnk&gl=us protein gel electrophoresis tips and troubleshooting guide]) | |||
* Carefully layer 50%EtOH 50% ddH2O on top of the gel to prevent the top of the gel from drying out | * Carefully layer 50%EtOH 50% ddH2O on top of the gel to prevent the top of the gel from drying out | ||
* | ** This also keeps the gel under anaerobic conditions, necessary for polymerization. | ||
* Store at 4 | ** It is very possible to ruin the desired evenness of the resolving gel by squirting this mixture in too fast. Janet has done it! | ||
* Wait 40 minutes for anaerobic polymerization. | |||
* Rinse ethanol/water mix off or you will get bubbles in the gel. | |||
** Rinse the gel aggressively by flushing with tap water from the sink. | |||
** Rinse several times with ddH2O | |||
** Prop the gel against the casting stand on its side so the liquid drains to one edge. Remove this with paper towel. | |||
* Store at 4 <sup>o</sup>C wrapped in a wet paper towel and saran wrap if you're not going to use it right away. | |||
** You can also store in the fridge after pouring the stacking gel. | |||
=== '''Pour the Stacking Gel'''=== | === '''Pour the Stacking Gel'''=== | ||
* | * Place gel in gel holder & ensure no water is sitting on top of the resolving gel. | ||
* | ** Remove liquid on top if you didn't already do so. Removal of ethanol/water or butanol is important to prevent bubbles. | ||
* | ** Dry surface of gel carefully with Kimwipe or paper towel | ||
* Dry surface of gel carefully with Kimwipe or paper towel | |||
** It can be a little gooey | ** It can be a little gooey | ||
* Mix components of the amounts in the Gel Mix link. Mix in the order listed. | * Mix components of the amounts in the Gel Mix link. Mix in the order listed. | ||
**[http://mach7.bluehill.com/proteinc/tutorial/sdspage.html Gel Mix Recipe] | **[http://mach7.bluehill.com/proteinc/tutorial/sdspage.html Gel Mix Recipe] | ||
** Don't add APS/TEMED until ready to pour | ** Don't add APS/TEMED until ready to pour | ||
* Use pipette to put gel mix into the gap between the plates | * Use pipette to put gel mix into the gap between the plates. | ||
* Insert the comb being 'careful not to trap any bubbles' | ** Filling to the top reduces the probability of trapping bubbles under the comb when you insert the comb. | ||
** It is much easier to avoid bubbles if you fill the space with enough solution that it spills over the top as you put the comb in. | * Insert the comb being '''careful not to trap any bubbles''' | ||
* Attach binder clips to help hold the comb in while drying. One | ** It is much easier to avoid bubbles if you fill the space with enough solution that it spills over the top as you put the comb in. It also helps to lower one side completely, then lower the other side down. | ||
* Leave for | * Attach 2 1" wide binder clips to each gel to help hold the comb in while drying. One on either side of the casting stand clamp. | ||
* | ** This will help ensure the edge wells don't get ruined -[[User:Janet_B._Matsen|JM 1/2013]] | ||
** On 1/3/2012 [[User:Janet_B._Matsen|JM]] didn't use binder clips and lost an edge well on two out of 6 gels, which usually doesn't happen. | |||
* Leave for 30 minutes while polymerization occurs. | |||
=== '''Wrapping up & Storing'''=== | |||
* Put binder clips away | |||
* Rinse everything off | |||
** Rinse gel under water with comb in to wash away solution that spilled over. | |||
** Remove comb from gel carefully (don't want to slant the parts that stick up) & soak in soapy water | |||
** Rinse casting stand, & rub the bottom where acrylamide tends to dry on | |||
** rub fingers over the comb to remove crust, rinse, and put on drying rack. | |||
* Wrap gels in a wet paper towel & store in a plastic bag or shrink wrap. | |||
** You can store for a few weeks in the fridge (>= 6 weeks). Leave comb in (fine to remove comb, fill wells with water ALS 10-24-13), and wrap in a wet paper towel and cling wrap or a plastic baggie. | |||
[[image:binder_clips_protein_gel.jpg|thumb|center|binder clips squeeze the glass to the comb. Put them as far down as they go.]] | [[image:binder_clips_protein_gel.jpg|thumb|center|binder clips squeeze the glass to the comb. Put them as far down as they go.]] | ||
| Line 43: | Line 71: | ||
[[image:SDS-PAGE casting procedure.jpg|thumb|upright=3.0|center|SDS casting protocol]] | [[image:SDS-PAGE casting procedure.jpg|thumb|upright=3.0|center|SDS casting protocol]] | ||
=='''Sample Prep'''== | =='''Sample Prep & Gel Loading'''== | ||
===Sample Prep === | |||
==== Prepping from E. coli liquid culture ==== | |||
* If you are boiling cultures and adding total protein, you should know that TB buffer when mixed with loading dye and boild results in a lot of white precipitate. You can leave the TB in the sample if you want because the precipitate doesn't matter in SDS-PAGE as the gel below shows. | |||
** Experiment that proves TB causes white precipitate: [[image:2014_03_03 precipitate in SDS-PAGE TB culture samples.png|thumb|upright=2.5|center|100uL aliquots of the same overnight E. coli culture grown in [[Lidstrom:TB|TB medium]] were spun for ~3 minutes to pellet the cells. The TB media was left, replaced with water, or half was removed and replaced by water. Pellets were resuspended, and either home-made 4X Laemelli buffer or commercial Bio-Rad buffer was used. Cell suspensions were boiled for 5 minutes then photographed.]] | |||
=== Does that white precipitate after boiling matter? === | |||
* The picture above shows some frightening looking white precipitate. When these mixtures were run in a gel (after vortexing so lots of the white particulate was loaded), all samples ran the same: [[image:2014_03_07 precipitate in SDS-PAGE TB culture samples doesnt matter for gels .png|thumb|upright=2.5|center|white precipitate formed during boiling TB culture with 4X loading buffer doesn't affect the way the gel runs.]] | |||
=== How much to load === | |||
==== Total Cell Protein ==== | |||
* use 5-8 or even up to 20 ug protein per well. (for Mini Protean with 10 well comb) | * use 5-8 or even up to 20 ug protein per well. (for Mini Protean with 10 well comb) | ||
** Amanda runs 15 ug in skinny comb wells 2013/10/25 | |||
* 7.5 uL of turbid TB E. coli is about right for skinny wells. This becomes 10uL after adding 4X buffer. | |||
** [[image:2014_10_01 sample uLOD loadings of ACS 2P2F.png|thumb|upright=1.4|center|sample of loading different uL*OD amounts of total BL21(DE3 culture. Protein = His*6 tagged [http://www.rcsb.org/pdb/explore.do?structureId=2p2f ACS 2P2F]]] | |||
==== Purified Protein ==== | |||
* ~40 ng seems about right. -[[Users:Janet B. Matsen|JM]] | |||
** This means the minimum concentration is ~5ng/uL. When you load 7.5uL in the skinny-comb gels (becomes 10uL after adding 4X buffer) you get ~40 ng in the gel. | |||
* 4 to 8 uL*uM works well for ~50-70kDa enzymes. If the stock is 1uM, add 4-8uL. If it is 4uM, add 1-2uL. *'''[[User:Janet B. Matsen|JM]] 20 August 2014''' | |||
** [[image:2014_10_01 sample uLuM loadings of ACS 2P2F.png|thumb|upright=1.4|center|sample of loading different uL*uM amounts of protein. Protein = His*6 tagged [http://www.rcsb.org/pdb/explore.do?structureId=2p2f ACS 2P2F] ]] | |||
==== In general ==== | |||
* To avoid edge effects, add 1x sample buffer to unused wells. (from [http://webcache.googleusercontent.com/search?q=cache:SWfzFRUgWxUJ:https://www.lablife.org/g?a%3Divy_file%26fh%3DYXvc5936lyS6GTqz6vTO1JNeQgY-%26v%3D1%26id%3Dg2441.CgrzyhkmAUhlRXEHPWLRnldxQuk-%26f%3Dattachments+&cd=7&hl=en&ct=clnk&gl=us protein gel electrophoresis tips and troubleshooting guide]) | |||
** [[User:Janet B. Matsen|JM] doesn't do this 2014/3 | |||
* If biomass is not limiting, prep a significant excess of protein, which will allow you to re-run if your gel turns out poorly for any reason. | * If biomass is not limiting, prep a significant excess of protein, which will allow you to re-run if your gel turns out poorly for any reason. | ||
* '''Maximum volumes''': | |||
** [[image:2014 01 14 maximum volumes for BioRad SDS-PAGE.png|thumb|upright=1.4|center|Maximum loading volumes for BioRad gels ([http://www.bio-rad.com/webroot/web/pdf/lsr/literature/Bulletin_6040A.pdf source]). As of 1/2014, the Lidstrom lab uses Mini-Format Gels with either 8 or 15 wells, so the maximum volumes are 30 or 15uL. Be careful not to let the samples splash into adjacent wells!]] | |||
** Load 12-15 uL, absolute max is 30 uL for 10-well comb in mini-protean | |||
*** '''Note: the amount you load affects the apparent size of the band''': [[image:2014 01 10- loading of gel affects apparent band size.png|thumb|upright=2.0|center|The amount of protein loaded in an SDS-PAGE lane affects the apparent band size. Notice the difference between 6uL and 10 uL of the BL21(DE3) total protein lysates. The protein of interest is marked with a white dot/star]] | |||
* If gel is overloaded or under loaded, run a new gel with a different amount of dyed & boiled lysate | |||
=== How to prepare samples === | |||
*If protein normalizing: | *If protein normalizing: | ||
** lyse cultures by sonication. Use 20 1 second pulses while tube is in ice, let the samples rest 5 minutes on ice, then sonicate again. Wipe sonicator stick between uses. Don't touch the tip while it is on. | ** lyse cultures by sonication. Use 20 1 second pulses while tube is in ice, let the samples rest 5 minutes on ice, then sonicate again. Wipe sonicator stick between uses. Don't touch the tip while it is on. | ||
| Line 64: | Line 123: | ||
***c) 37 degrees for 30 minutes (Membrane proteins or others that do not enter the gel otherwise may benefit from this type of sample preparation) | ***c) 37 degrees for 30 minutes (Membrane proteins or others that do not enter the gel otherwise may benefit from this type of sample preparation) | ||
** consider centrifugation to remove insoluble?? | ** consider centrifugation to remove insoluble?? | ||
** heat sample to | ** heat sample to 37<sup>o</sup>C to solubilize precipitated SDS?? | ||
=== Consolidated/Pictorial Protocol === | === Consolidated/Pictorial Protocol === | ||
[[image:SDS-PAGE sample prep and loading.jpg|thumb|upright=3.0|center|SDS-PAGE sample prep and loading]] | [[image:SDS-PAGE sample prep and loading.jpg|thumb|upright=3.0|center|SDS-PAGE sample prep and loading]] | ||
| Line 74: | Line 131: | ||
*Bio-Rad Mini-Cell Setup | *Bio-Rad Mini-Cell Setup | ||
**If only 1 gel, use buffer dam to replace second gel | **If only 1 gel, use buffer dam to replace second gel | ||
* | *'''Position the gels with the shorter plate facing inward!''' | ||
* Apply pressure on gel holder and gels as you close the tabs to seal the center compartment. | * Apply pressure on gel holder and gels as you close the tabs to seal the center compartment. | ||
[[image:gel holder.jpg|thumb|center|Mini-cell Gel holder]] | [[image:gel holder.jpg|thumb|center|Mini-cell Gel holder]] | ||
* Fill central compartment with running buffer | * Fill central compartment with running buffer | ||
** should fill sample wells | ** should fill sample wells | ||
* Pour | * Pour more into the outer compartment to specified line | ||
* Load gel | * Load gel | ||
** Make sure you will be able to determine the orientation of your gel after it is stained. Asymmetry is good! | ** Make sure you will be able to determine the orientation of your gel after it is stained. Asymmetry is good! | ||
* Make sure to color/charge-match the cords to the power unit as the electrodes in the gel holder to the contacts in the lid. | * Make sure to color/charge-match the cords to the power unit as the electrodes in the gel holder to the contacts in the lid. | ||
* Run @ 60 V for | * Run @ 60 V for ~15 min, then 200 V for ~ 20+ min. | ||
** Note: ladder looks blurry while running through the stacking gel; don't be alarmed unless it still looks blurry in the resolving gel. | ** Note: ladder looks blurry while running through the stacking gel; don't be alarmed unless it still looks blurry in the resolving gel. | ||
**Can skip the 60V step if you don't need a gorgeous gel | **Can skip the 60V step if you don't need a gorgeous gel | ||
** Amanda runs 20 min at 200V, then checks frequently to make sure the protein doesn't run off the gel. | ** Amanda runs 20 min at 200V, then checks frequently to make sure the protein doesn't run off the gel. | ||
** Lower voltages can be considered if 200V leads to poor resolution. | |||
*** "[the] best typical conditions for electrophoresis are at 10-15 V/cm." (reference: [http://webcache.googleusercontent.com/search?q=cache:SWfzFRUgWxUJ:https://www.lablife.org/g?a%3Divy_file%26fh%3DYXvc5936lyS6GTqz6vTO1JNeQgY-%26v%3D1%26id%3Dg2441.CgrzyhkmAUhlRXEHPWLRnldxQuk-%26f%3Dattachments+&cd=7&hl=en&ct=clnk&gl=us protein gel electrophoresis tips and troubleshooting guide]) | |||
==Storing Samples After Use== | |||
* Amanda was taught to flash-freeze (liquid N<sub>2</sub>) samples and store them at -80<sup>o</sup>C. Janet loves liquid N2 (particularly dumping it on the ground after use) so I follow along without question. | |||
* Ladder is stored at -80oC. An aliquot of PageRuler Prestained that sat out at room temp for 1 week looked perfect in a gel, so don't worry about that ladder's stability. | |||
== '''Staining, Destaining, & Visualization''' == | == '''Staining, Destaining, & Visualization''' == | ||
=== Old school bromphenol blue dye === | |||
* dye overnight or cycles of 1 min @ power 6 in the microwave | * dye overnight or cycles of 1 min @ power 6 in the microwave | ||
** microwave by Bo's bench. Let it vent a little in the hood between heating events. | ** microwave by Bo's bench. Let it vent a little in the hood between heating events. | ||
* Return dye to container | * Return dye to container | ||
*Rinse to remove residual dye | * Rinse to remove residual dye | ||
*Destain (I do | * Destain (2 - 4 cycles) | ||
** The destain we use is just as potent when diluted 1:1 with tap water. I do this to save material and destain solution mixing time. -[[User:Janet_B._Matsen|JM 12/2012]] | |||
** Save the destain from the 3rd & 4th cycles, and re-use it for a future gel's 1st destain. | |||
* Image with white light transillumination | |||
** use the white light box, set the Gel Logic program to "white light transillumination", and use an exposure time of about 0.15 seconds. | |||
*** Using 1 second (and a tighter aperture) results in a blurry image | |||
*** Using less than ~ 0.15 seconds leads to inconsistent exposures. (The final image won't usually match the exposure level shown in the preview screen.) | |||
[[image:two exposures of one SDS gel.jpg|thumb|upright=3.0|center|two exposures of one SDS gel - the one on the right is better (~0.15 second exposure)]] | |||
=== New school GelCode Blue Safe Protein Stain === | |||
* Product Info: | |||
** [http://www.piercenet.com/product/gelcode-blue-safe-protein-stain GelCode Blue Safe Protein Stain]: A coomassie gel stain that's safer and less costly to ship. | |||
** Pierce Item #1860957, Item# 24594 | |||
* How to use: | |||
** Wash gel 3x in tap water (says Frances). Let it marinate ~10 min each time. | |||
** Pour enough on to cover gel | |||
** stain 30 min - overnight. (Frances likes overnight) | |||
** Dump leftover stain down drain | |||
*** you might consider re-using bugger if your bands are fat | |||
** Destain in water, but not overnight (too long in Amanda's experience) | |||
== '''Other Resources''' == | == '''Other Resources''' == | ||
| Line 101: | Line 183: | ||
*[http://www.bio-rad.com/webroot/web/pdf/lsr/literature/4110182D.PDF Instructions for BioRad Rainbow Std] | *[http://www.bio-rad.com/webroot/web/pdf/lsr/literature/4110182D.PDF Instructions for BioRad Rainbow Std] | ||
*[http://www.bio-rad.com/webroot/web/pdf/lsr/literature/10007296.PDF Mini-PROTEAN® Tetra Cell manual] | *[http://www.bio-rad.com/webroot/web/pdf/lsr/literature/10007296.PDF Mini-PROTEAN® Tetra Cell manual] | ||
* more comprehensive [http://www.bio-rad.com/webroot/web/pdf/lsr/literature/Bulletin_6040A.pdf BioRad manual] for SDS-PAGE ([[User:Janet B. Matsen|Janet]]'s favorite) | * more comprehensive [http://www.bio-rad.com/webroot/web/pdf/lsr/literature/Bulletin_6040A.pdf BioRad manual] for SDS-PAGE ([[User:Janet B. Matsen|Janet]]'s favorite) | ||
* [http://www.bio-rad.com/webroot/web/pdf/lsr/literature/Bulletin_4110001E.pdf Criterion manual] (for larger sized gels) | * [http://www.bio-rad.com/webroot/web/pdf/lsr/literature/Bulletin_4110001E.pdf Criterion manual] (for larger sized gels) | ||
* [https://sites.google.com/site/sdspageprotocol/pouring-a-gel video how-to] (not vetted) | * [https://sites.google.com/site/sdspageprotocol/pouring-a-gel video how-to] (not vetted) | ||
* [https://maps.google.com/maps?saddr=latona+pub&daddr=530+Broadway+E,+Seattle,+WA&hl=en&ll=47.649894,-122.318687&spn=0.09656,0.222988&geocode=Fax51wIdu3W1-CGqpyQuHP3hZymNDlZQaRSQVDGqpyQuHP3hZw%3BFdSu1gIdDoq1-CnJWB6ILhWQVDEMTNB7f44wVQ&oq=530+broad&dirflg=r&ttype=now&noexp=0&noal=0&sort=def&mra=ltm&t=m&z=13&start=0 article] about polymerization from BioRad | |||
== '''Mistakes to Be Careful About''' == | == '''Mistakes to Be Careful About''' == | ||
* using an "old" APS solution when making gels. The 10% weight/volume APS | === Prepping Gels === | ||
* using an "old" APS solution when making gels. The 10% weight/volume APS can be made fresh each time for best results. Don't use a solution that is more than a month or so old. The gels won't polymerize. | |||
* not mixing the liquid gel mixture enough | * not mixing the liquid gel mixture enough | ||
** Janet pipettes up and down with a 10 or 15 mL pipette about 10 times, being careful not to bubble the solution. | |||
* letting the gel dry too long after pouring the stacking gel (comb step) | * letting the gel dry too long after pouring the stacking gel (comb step) | ||
** the very edges can shrivel up, which becomes a problem when you try to use those edge lanes | ** the very edges can shrivel up, which becomes a problem when you try to use those edge lanes | ||
*taking the gel off the casting stand before it has polymerized | *taking the gel off the casting stand before it has polymerized | ||
** entire sample will leak out | ** entire sample will leak out | ||
* | |||
* using too much beta-mercaptoethanol in your | *If using commercially prepared gels, don't forget to remove the tape on the bottom of the gel! If you load your samples and turn on the power and see voltage but no current, check for tape removal (duh, Nicole)! | ||
=== Preparing Samples === | |||
* using too much beta-mercaptoethanol in your sample buffer | |||
** should have < 1% beta-mercaptoethanol in the mix after you add sample buffer to the | ** should have < 1% beta-mercaptoethanol in the mix after you add sample buffer to the | ||
** too much reduction of cysteines is bad: will alter structure and even cleave proteins. | ** too much reduction of cysteines is bad: will alter structure and even cleave proteins. | ||
=== Running Gels === | |||
* sample sloshing out of the well you are trying to load and falling into a neighboring well | |||
* Electrode buffer leaking out from between the two plates | |||
** Gel will run very unusually or stop running. Keep an eye out! | |||
** avoid by making '''sure''' the plates are well-seated | |||
* Using Tris-HCl instead of Tris in the electrode buffer | |||
* not having the lid to the running unit on all the way. --> poor electrical contact & blurry bands | * not having the lid to the running unit on all the way. --> poor electrical contact & blurry bands | ||
* | * Using resolving gel Tris Buffer instead of the intended electrode buffer. | ||
** | ** The results of said mistake: [[User:Janet B. Matsen|JM 2013/01/04]] [[image:SDS-PAGE gel run with resolving gel buffer instead of the proper electrode buffer.jpg|thumb|upright=1.5|center|SDS-PAGE gel run with resolving gel buffer instead of the proper electrode buffer]] | ||
** | |||
=== Visualizing Gels === | |||
* Gels tear easily when warm. | |||
* Be sure not to tear the gel as you take it out. It is good to wet the glass after you remove the cover glass plate. | |||
== '''Recipes'''== | == '''Recipes'''== | ||
All recipes except the staining & destaining solution are from the [http://www.bio-rad.com/webroot/web/pdf/lsr/literature/10007296.PDF Mini-PROTEAN® Tetra Cell manual] | All recipes except the staining & destaining solution are from the [http://www.bio-rad.com/webroot/web/pdf/lsr/literature/10007296.PDF Mini-PROTEAN® Tetra Cell manual] | ||
=== 10% (w/v) APS === | |||
* 0.10 g/1 mL diH2O = 100 mg/mL (prepare fresh daily for best results; most people do every few weeks.) ([http://www.bio-rad.com/webroot/web/pdf/lsr/literature/Bulletin_6040A.pdf link]) | |||
=== Loading Buffer === | === Loading Buffer === | ||
* | * Mix pre-made concentrated loading buffer with fresh beta-mercaptoethanol prior to each use. | ||
** BioRad's loading buffer recipe: 3.55 mL deionized water, 1.25 mL 0.5 M Tris-HCl pH 6.8, 2.5 mL glycerol, 2.0 mL of 10% (w/v) SDS, 0.2 mL of 0.5% (w/v) Bromophenol Blue. Total volume = 9.5 mL. | ** [http://wolfson.huji.ac.il/purification/PDF/Tag_Protein_Purification/Ni-NTA/Clontech_Talon_protocol.pdf TALON recipe]: 5X SDS PAGE Sample Buffer | ||
*** 15% β-Mercaptoethanol (β-ME) | |||
*** 15% SDS | |||
*** 50% Glycerol | |||
*** 1.5% Bromophenol blue | |||
** BioRad's loading buffer recipe: | |||
*** 3.55 mL deionized water, 1.25 mL 0.5 M Tris-HCl pH 6.8, 2.5 mL glycerol, 2.0 mL of 10% (w/v) SDS, 0.2 mL of 0.5% (w/v) Bromophenol Blue. Total volume = 9.5 mL. | |||
* mix 50 uL beta-mercaptoethanol to 950 uL sample buffer prior to use. | * mix 50 uL beta-mercaptoethanol to 950 uL sample buffer prior to use. | ||
** Scaled back 5x: 10 uL beta-mercaptoethanol + 190 uL 5x buffer | ** Scaled back 5x: 10 uL beta-mercaptoethanol + 190 uL 5x buffer | ||
| Line 134: | Line 239: | ||
** Janet presumes this is 1 part sample per 2 parts buffer, but isn't sure (???) | ** Janet presumes this is 1 part sample per 2 parts buffer, but isn't sure (???) | ||
*Amanda's recipe is a little different: 4x mix made of: 4 mL glycerol, 0.8 g SDS, 2.5 mL 1M Tris-HCl pH 6.8, 80 uL of bromophenol blue slurry (5 mg/mL = 0.5% (w/v)), H2O to 8 mL. | *Amanda's recipe is a little different: 4x mix made of: 4 mL glycerol, 0.8 g SDS, 2.5 mL 1M Tris-HCl pH 6.8, 80 uL of bromophenol blue slurry (5 mg/mL = 0.5% (w/v)), H2O to 8 mL. | ||
** Amanda mixes 160 uL | ** Amanda mixes 160 uL 4X buffer, 8 uL beta-mercapto-ethanol, 32 uL H2O. Use 10 uL per 25 uL of cells. [https://docs.google.com/spreadsheet/ccc?key=0AlVxrZi130nMdEJmX1hqRGZPREtWVVQ0Q21kR0stdkE Recipe Comparison (to BioRad's)] | ||
* Can add up to 8M urea for really hydrophobic proteins | * Can add up to 8M urea for really hydrophobic proteins | ||
=== Running Buffer: === | === Running/Electrode Buffer: === | ||
* 10x SDS | * 10x SDS-PAGE (1 L) (BioRad catalog #161-0732) = 250 mM Tris, 1.92 M glycine, 1% SDS, pH 8.3 | ||
**Mix: | |||
***Tris base 30.30 g '''DO NOT USE Tris-HCl''' | |||
***Glycine 144.10 g | |||
***SDS 10.00 g | |||
***diH2O to 1 L | |||
**'''Do not adjust the pH (~pH 8.3)''' | |||
* Make 1L of 1x for use. | * Make 1L of 1x for use. | ||
*Store at 4oC. | *Store at 4oC. | ||
* This buffer is used while running proteins through the gel. Pour it in as the instructions for the box explain. Pour back into bottle for re-use afterward. | * This buffer is used while running proteins through the gel. Pour it in as the instructions for the box explain. Pour back into bottle for re-use afterward. | ||
* | * It is best practice to not re-use electrode buffer. Our lab does, however, re-use the buffer ~ 4 (but sometimes up to 10) times. | ||
=== Staining Buffer (Coomassie Brilliant Blue G-250): === | === Staining Buffer (Coomassie Brilliant Blue G-250): === | ||
| Line 153: | Line 264: | ||
* 30% methanol, 10% acetic acid, water | * 30% methanol, 10% acetic acid, water | ||
* some labs use much less methanol & acetic acid; some use plain water. | * some labs use much less methanol & acetic acid; some use plain water. | ||
* Janet rinses in plain water before using our | * Janet rinses in plain water before using our destaining buffer. | ||
== '''Acrylamide toxicity''' == | == '''Acrylamide toxicity''' == | ||
*[http://en.wikipedia.org/wiki/Acrylamide Acrylamide] is [http://oehha.ca.gov/prop65/acrylamideqa.html toxic] to your nervous system, and may be a carcinogen. The unpolymerized form is toxic, but the polymerized form is much less toxic. ALWAYS wear gloves and wipe up spills - once the solution | *[http://en.wikipedia.org/wiki/Acrylamide Acrylamide] is [http://oehha.ca.gov/prop65/acrylamideqa.html toxic] to your nervous system, and may be a carcinogen. The unpolymerized form is toxic, but the polymerized form is much less toxic. ALWAYS wear gloves and wipe up spills - once the solution dries, the dust can be [http://www.nap.edu/openbook.php?record_id=4911&page=250 inhaled]. Interestingly, fried starchy/sugary foods naturally contain acrylamide, too. | ||
*more than you want to know about acrylamide toxicity can be found [http://jifsan.umd.edu/docs/acrylamide2002/wg4_toxicology_bg.pdf here] | *more than you want to know about acrylamide toxicity can be found [http://jifsan.umd.edu/docs/acrylamide2002/wg4_toxicology_bg.pdf here] | ||
== Ladder == | == Ladder == | ||
The Lidstrom Lab PageRuler ladder best. ([https://tools.lifetechnologies.com/content/sfs/manuals/MAN0011772_PgRuler_Prestain_Protein_Lad_UG.pdf manual]) | |||
[[image:PageRuler Protein Gel Legend.jpg|thumb|center|PageRuler protein gel legend]] | [[image:PageRuler Protein Gel Legend.jpg|thumb|center|PageRuler protein gel legend]] | ||
Notes: | |||
* The mobility of prestained proteins can vary in different SDS-PAGE buffer systems ([https://tools.lifetechnologies.com/content/sfs/manuals/MAN0011772_PgRuler_Prestain_Protein_Lad_UG.pdf manual]) | |||
* We load 4uL per well for 0.75mm thick gels; the manual suggests 5uL. | |||
* You can't get use the ladders to approximate/quantify how much protein you added: | |||
** "Thermo Scientific ladders are not designed for protein quantification. For quantification, we would recommend to use a protein of known concentration as a reference." | |||
Other ladders: | Other ladders: | ||
| Line 167: | Line 285: | ||
== Combs/Loading Volumes == | == Combs/Loading Volumes == | ||
* We currently only have the mini-protean gel running boxes. -[[User:Janet B. Matsen|JM]] 10/2012 | * We currently only have the mini-protean gel running boxes. -[[User:Janet B. Matsen|JM]] 10/2012 | ||
[[image:SDS combs and loading volumes.jpg|thumb|center|SDS combs and loading volumes]] | [[image:SDS combs and loading volumes.jpg|upright=3.0|thumb|center|SDS combs and loading volumes]] | ||
== Troubleshooting == | |||
* [http://www.hycultbiotech.com/media/wysiwyg/Troubleshooting_SDS-PAGE_1.pdf Hycult Biotech] troubleshooting guide | |||
* [http://webcache.googleusercontent.com/search?q=cache:SWfzFRUgWxUJ:https://www.lablife.org/g?a%3Divy_file%26fh%3DYXvc5936lyS6GTqz6vTO1JNeQgY-%26v%3D1%26id%3Dg2441.CgrzyhkmAUhlRXEHPWLRnldxQuk-%26f%3Dattachments+&cd=7&hl=en&ct=clnk&gl=us protein gel electrophoresis tips and troubleshooting guide] includes troubleshooting after general tips | |||
* [http://www.hycultbiotech.com/media/wysiwyg/Troubleshooting_SDS-PAGE_1.pdf nippongenetics guide] | |||
==Questions/Answers/Facts== | ==Questions/Answers/Facts== | ||
===What do all of the reagents do? === | ===How do you prepare soluble and insoluble fractions for SDS-PAGE?=== | ||
Yakov Kipnis of the Baker lab wrote in 11/2014: | |||
"Soluble and insoluble fractions of cell lysate look distinct on gel (ratios of band intensities for various proteins is quite different), therefore if you see large portion of unlysed cells contaminating insoluble fraction distinction between soluble/insoluble becomes less pronounced. To make the comparison easier I typically run 3 lanes/sample (total cell lysate/soluble(sup after spin)/insoluble(pellet after spin) I always try to load similar amount of protein on a gel. For "total" and "soluble" it is straightforward, to help solubilize as much of "insoluble" as possible I resuspend pellet in small volume of 8-9 M urea (avoid GuHCl it destroys SDS gels) first and reconstitute to the same volume before taking small aliquot for gel sample. | |||
Example: total volume of the lysate 500 uL (take 10 uL for gel="total"), spin (take 10 uL="soluble"), remove supernatant as much as possible, add 20-50 uL 9M urea, resuspend pellet, dilute to 490 uL by water/buffer (take 10 uL="insoluble"). This way theoretically overlay of "soluble" + "insoluble" lanes should give you "total", if corresponding bands do not sum up there is a chance something unaccounted happened. Later you can skip "total" to save space on gel." | |||
===What do all of the reagents do? How long do they last? === | |||
* SDS | * SDS | ||
**In loading buffer and often in gels (not necessary to include in gels; can be used in sample buffer) | **In loading buffer and often in gels (not necessary to include in gels; can be used in sample buffer) | ||
* APS & TEMED | * APS & TEMED | ||
** polymerize acrylamide | ** polymerize acrylamide | ||
**Stability: | |||
*** Biorad customer service 10/2012: "TEMED breaks down through interaction with oxygen, which produces many products in small amounts. TEMED, if stored properly, excluding oxygen, should not break down. It can pool up under the liner of the cap, and since it has very low viscosity, can creep between the threads of the cap and those of the bottle. '''TEMED color is a good indicator of degradation. TEMED becomes a darker yellow color when degraded.''' Our APS is guaranteed for 1 year at room temperature in dry form (powder). APS should last indefinitely if stored desiccated. " | |||
***brand new BioRad TEMED opened 11/2012 was clear, not yellow. The old bottle was clear-yellow. | |||
* glycine | * glycine | ||
** carry charge in the opposite direction as the negatively-charged SDS-covered proteins | ** carry charge in the opposite direction as the negatively-charged SDS-covered proteins | ||
| Line 181: | Line 312: | ||
* glycerol in loading buffer: helps sample sink | * glycerol in loading buffer: helps sample sink | ||
* Bromphenol Blue | * Bromphenol Blue | ||
=== All about the "stacking" layer of the gel === | |||
* The stacking layer originally was a lower density acrylamide with a lower pH. The goal of "stacking" was to gather all the proteins into a tight band so that they all start from the "starting line" at the same time, and appear more clearly after completion. The lower pH of the stacking gel is intended to affect how much the glycine in the running buffer is attracted to the proteins, which affects the proteins mobility. [http://sdspage.homestead.com/ This video] explains it if you can tolerate the corniness. | |||
* There are, however, different ways to achieve stacking: | |||
** lower density gel on top that isn't at a different pH | |||
*** Example: [http://www.bio-rad.com/webroot/web/pdf/lsr/literature/1658100C.pdf commercially available gels from BioRad]. The fixed-percentage gels have only one buffer ingredient. | |||
*** The fact that having different pH values in the stacking and resolving gels isn't so important clarifies why home-made gels that initially have different pHs in the two layers still work fine weeks after they were poured when presumably the buffers converge to equilibrium. | |||
** a gradient of acrylamide percentages | |||
*** A stacking gel is not necessary when using a gradient gel, as the gradient itself performs this function. ([http://www.piercenet.com/method/overview-electrophoresis#precastgels thermo scientific]) | |||
=== Is it important to degas my water + buffer + acrylamide mix before adding the APS and TEMED, as the manuals recommend?=== | === Is it important to degas my water + buffer + acrylamide mix before adding the APS and TEMED, as the manuals recommend?=== | ||
* "We recommend that you degas the solutions to get rid of nitrogen. Sometimes, the nitrogen may come out of solution and form bubbles in your gels. " -BioRad customer support 10/26/2012 [[User:Janet B. Matsen|JM]] | * "We recommend that you degas the solutions to get rid of nitrogen. Sometimes, the nitrogen may come out of solution and form bubbles in your gels. " -BioRad customer support 10/26/2012 [[User:Janet B. Matsen|JM]] | ||
| Line 187: | Line 328: | ||
** Also note this is not the justification customer support provided. (see above) | ** Also note this is not the justification customer support provided. (see above) | ||
=== Why shouldn't we overlay the gels with butanol or | === Why shouldn't we overlay the gels with butanol or isopropanol as they polymerize? === | ||
*"We do not recommend using butanol/isopropanol because these may degrade the glue on the spacer plates and the plastic of the casting frame." -BioRad customer support 10/26/2012 [[User:Janet B. Matsen|JM]] | *"We do not recommend using butanol/isopropanol because these may degrade the glue on the spacer plates and the plastic of the casting frame." -BioRad customer support 10/26/2012 [[User:Janet B. Matsen|JM]] | ||
=== Should I soak my gel or run them | === Should I soak my gel or run them without samples before use? === | ||
* They say no: "I have not heard that soaking the gels in water will improve the run. Talking with my colleagues, we thought it would have been detrimental because the tris and chloride ions will diffuse out during this time and in theory, should have made your gel run poorly." | * They say no: "I have not heard that soaking the gels in water will improve the run. Talking with my colleagues, we thought it would have been detrimental because the tris and chloride ions will diffuse out during this time and in theory, should have made your gel run poorly." | ||
* | * We had an episode in October & November 2012 where the gels ran funny unless they were pre-run without samples, but this was resolved by replacing gel-pouring buffers someone made with commercial BioRad buffers. About the temporary issue: | ||
** Ladder (and all other protein samples) ran fine until 1/2 way through the gel, after which they nearly halted and stacked on top of one another. Immediately after, a ~1 cm wide band that spanned across the gel was visible as it refracted light differently. This band disappeared after a while of soaking, leading me to believe it was a buildup of one of the buffer compounds. When such a gel was soaked overnight in water and ladder was loaded in an unused adjacent well, and it ran perfectly without bunching up. This was a very repeatable phenomenon with all gels used before the problem was solved. | |||
* Potential problems with soaking or pre-running gels: | * Potential problems with soaking or pre-running gels: | ||
** You might ruin the [http://www.bio-rad.com/webroot/web/pdf/lsr/literature/Bulletin_6040A.pdf stacking nature] of the gel by | ** You might ruin the [http://www.bio-rad.com/webroot/web/pdf/lsr/literature/Bulletin_6040A.pdf stacking nature] of the gel by altering the buffer within the acrylamide matrix. This essentially converts your gel to a continuous buffer gel system, known to give more blurry bands. | ||
=== Is it ok to re-use my electrode buffer? === | === Is it ok to re-use my electrode buffer? === | ||
| Line 200: | Line 341: | ||
=== How long can I store my acrylamide gels? === | === How long can I store my acrylamide gels? === | ||
* "Tris-HCl resolving gels are prepared at pH 8.6–8.8. At this basic pH, polyacrylamide slowly hydrolyzes to polyacrylic acid, which can compromise separation. For this reason, Tris-HCl gels have a relatively short shelf life. In addition, the gel pH can rise to pH 9.5 during a run, causing proteins to undergo deamination and alkylation. This may diminish resolution and complicate | * "Tris-HCl resolving gels are prepared at pH 8.6–8.8. At this basic pH, polyacrylamide slowly hydrolyzes to polyacrylic acid, which can compromise separation. For this reason, Tris-HCl gels have a relatively short shelf life. In addition, the gel pH can rise to pH 9.5 during a run, causing proteins to undergo deamination and alkylation. This may diminish resolution and complicate post electrophoresis analysis." ([http://www.bio-rad.com/webroot/web/pdf/lsr/literature/Bulletin_6040A.pdf reference]) | ||
** note: | ** note: commercial Tris gels have a shelf life of 6 months to 1 year. Amanda & Ceci don't keep hand-poured gels for more than a few (~4) weeks. ([[User:Janet B. Matsen|JM]] 11/2012) | ||
*** [[users:Janet B. Matsen|JM]] uses gels that are up to 11 weeks old. They work perfectly. I haven't tested longer yet. | |||
** It is possible the commercial gels have other means of extending shelf life. | ** It is possible the commercial gels have other means of extending shelf life. | ||
=== | === What is the difference between Coomassie R and Coomassie G? === | ||
* [https://www.nationaldiagnostics.com/electrophoresis/article/staining-protein-gels-coomassie-blue useful link] | * [https://www.nationaldiagnostics.com/electrophoresis/article/staining-protein-gels-coomassie-blue useful link] | ||
* note: Coomassie Brilliant Blue G-250 differs from Coomassie Brilliant Blue R-250 by the addition of two methyl groups. We use the G form. Read more about the R form [http://en.wikipedia.org/wiki/Coomassie_Brilliant_Blue here]. | * note: Coomassie Brilliant Blue G-250 differs from Coomassie Brilliant Blue R-250 by the addition of two methyl groups. We use the G form. Read more about the R form [http://en.wikipedia.org/wiki/Coomassie_Brilliant_Blue here]. | ||
| Line 211: | Line 353: | ||
* The G-250 form the colloidal particles in an aqueous solution. This is an advantage for staining a gel because the colloids tend not to stain the gel matrices, reducing the background problem. When the colloids come close to the proteins, the dye molecule is removed from the colloids by the nearby proteins due to the higher affinity of proteins to the dye. | * The G-250 form the colloidal particles in an aqueous solution. This is an advantage for staining a gel because the colloids tend not to stain the gel matrices, reducing the background problem. When the colloids come close to the proteins, the dye molecule is removed from the colloids by the nearby proteins due to the higher affinity of proteins to the dye. | ||
* R-250, on the other hand, doesn't form the colloids. Rather, an individual dye molecule is dispersed in a solution. Therefore, the dye molecules can interact not only with proteins but with gel matrices freely, creating the background staining issue. | * R-250, on the other hand, doesn't form the colloids. Rather, an individual dye molecule is dispersed in a solution. Therefore, the dye molecules can interact not only with proteins but with gel matrices freely, creating the background staining issue. | ||
=== Should I throw my gel away if I forgot to add SDS to the resolving gel? === | |||
* Janet thinks no, for two reasons: | |||
** The manual says "Bio-Rad’s precast gels also do not contain SDS and can be used for native or denaturing PAGE." ([http://www.bio-rad.com/webroot/web/pdf/lsr/literature/Bulletin_6040A.pdf citation]) | |||
** In 11/2012 I forgot to, and the proteins ran wonderfully, anyway. | |||
== Other Tips == | == Other Tips == | ||
=== Collecting samples in PCR strip tubes makes life nice! === | |||
* You can concentrate samples using the PCR strip spinner on Marina's bench. '''It must be balanced!''' | |||
* You can do boiling-driven lysis in a thermocycler | |||
* Samples don't take up a lot of room in the freezer. | |||
* To write on them (for recording the date, and strip number if you have multiple) you can use a fine-tip sharpie and cover it with a ring of scotch-tape. Without tape, the writing tends to wear off. | |||
* We are in love with individually removable caps that stay attached as is sold [http://www.bulldog-bio.com/pcr_plastics.html here]. | |||
** Samples don't splattering when you take of the caps, reducing cross-contamination and mess | |||
** If you only have one cap open at a time when loading your gel, you can tell much more easily which sample you just loaded, reducing the possibility that you have a problem. | |||
** Do, still, take each cap off slowly when opening them if there is liquid in the lid of the chamber. Often after shaking the strip to mix it, I open the cap quickly and splurt the blue sample on my clothes. It always rinses clean after adding water, but it is nicer if this doesn't happen. | |||
=== Excess salt in SDS-PAGE samples causes fuzzy bands and narrowing of gel lanes toward the bottom of the gel === | === Excess salt in SDS-PAGE samples causes fuzzy bands and narrowing of gel lanes toward the bottom of the gel === | ||
* If the ionic strength is very high, no bands will appear in the lower part of the gel (a vertical streak will appear instead) and the dye front will be wavy instead of straight. Deionize any sample with a total ionic strength over 50 mM using columns such as Micro Bio-Spin™ columns, which contain 10 mM Tris at a pH suitable for SDS-PAGE. ([http://www.bio-rad.com/webroot/web/pdf/lsr/literature/Bulletin_6040A.pdf source]) | * If the ionic strength is very high, no bands will appear in the lower part of the gel (a vertical streak will appear instead) and the dye front will be wavy instead of straight. Deionize any sample with a total ionic strength over 50 mM using columns such as Micro Bio-Spin™ columns, which contain 10 mM Tris at a pH suitable for SDS-PAGE. ([http://www.bio-rad.com/webroot/web/pdf/lsr/literature/Bulletin_6040A.pdf source]) | ||
| Line 229: | Line 386: | ||
===KCl causes SDS to precipitate === | ===KCl causes SDS to precipitate === | ||
* If you samples contain KCl you should dilute them or methanol precipitate them and resuspend them in 1X sample buffer. With low concentrations of KCl (<200 mM) you can run them on the gel but you should | * If you samples contain KCl you should dilute them or methanol precipitate them and resuspend them in 1X sample buffer. With low concentrations of KCl (<200 mM) you can run them on the gel but you should load every lane with sample buffer containing the same concentration of KCl (even if they are blanks). This will help the gel run a little less anomalously. ([http://mullinslab.ucsf.edu/Protocols%20HTML/SDS_PAGE_protocol.htm source]) | ||
== Supplies == | |||
* BioRad sells most of the reagents we use. You can make most of them yourself, if you'd like. | |||
* The "diaper" that goes under the gel casting stand: | |||
** [[User:Janet B. Matsen|Janet]] called BioRad , UW EH&S, and VWR on 11/29/2012 to identify a special pad that is particularly good for absorbing liquid acrylamide mixtures used in pouring acrylamide gels. The idea is that such a pad would prevent the dried acrylamide from being re-released into the air and breathed by scientists. Unfortunately, this doesn't seem to exist. | |||
** We ordered [https://us.vwr.com/store/catalog/product.jsp?catalog_number=89126-794 these] | |||
Revision as of 12:11, 9 February 2015
Return: Lidstrom Lab Protocols
Return: official OpenWetWare SDS-PAGE
Contact Janet Matsen with questions, comments, & corrections.
Intro
Gel Prep
- Clean cover plate and thicker spacer plate (75 mM gap)
- Soap and Water: scrub with gloved fingers. Ensure all chunks are gone.
- Rinse with water, then ethanol. Prop up vertically against green clamps for stands so they drain and dry.
- Setup one spacer plate and one cover plate in each gel holder
- The cover plate goes on the side of the spacer plate with the spacers in order to create a small gap between the plates
- Put on the black counter top to pinch closed. Don't put it on the absorbent mat, as this slight unevenness will increase your gel leakage rate. Janet had ~33% of gels leak until she started doing it on the black counter without padding; now she has ~5-10% of poured gels leak.
- Put the gel holder into the casting stand
- Make a fresh 10% (w/vol) APS solution. Don't use an "old" solution; the gel won't polymerize. 10% w/v = 0.1 g/mL.
- 1 month old seems to be safe, but up to 3 months may be ok.
Pour the Resolving Gel
- Mix components of the amounts in the Gel Mix link for the resolving gel (Recipes is for 4 gels). Mix in the order listed.
- Gel Mix Recipe
- Don't add APS/TEMED until ready to pour
- The APS solution should be < 1 month old. Up to 2 months might be ok, but we don't have great data for this.
- A 3 month old solution failed to cause polymerization & the liquid spilled out from between the plates when released from the stand. -JM 10/2012
- The BioRad protocols insist you should make it fresh every time, but we never do if we have one made within the last 4 weeks
- Use pipette to put gel mix into the gap between the plates. Fill to about the bottom of the green line that is below the short plate.
- leave room for comb + 1 cm of resolving gel (citation: protein gel electrophoresis tips and troubleshooting guide)
- Carefully layer 50%EtOH 50% ddH2O on top of the gel to prevent the top of the gel from drying out
- This also keeps the gel under anaerobic conditions, necessary for polymerization.
- It is very possible to ruin the desired evenness of the resolving gel by squirting this mixture in too fast. Janet has done it!
- Wait 40 minutes for anaerobic polymerization.
- Rinse ethanol/water mix off or you will get bubbles in the gel.
- Rinse the gel aggressively by flushing with tap water from the sink.
- Rinse several times with ddH2O
- Prop the gel against the casting stand on its side so the liquid drains to one edge. Remove this with paper towel.
- Store at 4 oC wrapped in a wet paper towel and saran wrap if you're not going to use it right away.
- You can also store in the fridge after pouring the stacking gel.
Pour the Stacking Gel
- Place gel in gel holder & ensure no water is sitting on top of the resolving gel.
- Remove liquid on top if you didn't already do so. Removal of ethanol/water or butanol is important to prevent bubbles.
- Dry surface of gel carefully with Kimwipe or paper towel
- It can be a little gooey
- Mix components of the amounts in the Gel Mix link. Mix in the order listed.
- Gel Mix Recipe
- Don't add APS/TEMED until ready to pour
- Use pipette to put gel mix into the gap between the plates.
- Filling to the top reduces the probability of trapping bubbles under the comb when you insert the comb.
- Insert the comb being careful not to trap any bubbles
- It is much easier to avoid bubbles if you fill the space with enough solution that it spills over the top as you put the comb in. It also helps to lower one side completely, then lower the other side down.
- Attach 2 1" wide binder clips to each gel to help hold the comb in while drying. One on either side of the casting stand clamp.
- Leave for 30 minutes while polymerization occurs.
Wrapping up & Storing
- Put binder clips away
- Rinse everything off
- Rinse gel under water with comb in to wash away solution that spilled over.
- Remove comb from gel carefully (don't want to slant the parts that stick up) & soak in soapy water
- Rinse casting stand, & rub the bottom where acrylamide tends to dry on
- rub fingers over the comb to remove crust, rinse, and put on drying rack.
- Wrap gels in a wet paper towel & store in a plastic bag or shrink wrap.
- You can store for a few weeks in the fridge (>= 6 weeks). Leave comb in (fine to remove comb, fill wells with water ALS 10-24-13), and wrap in a wet paper towel and cling wrap or a plastic baggie.

Consolidated/Pictorial Protocol

Sample Prep & Gel Loading
Sample Prep
Prepping from E. coli liquid culture
- If you are boiling cultures and adding total protein, you should know that TB buffer when mixed with loading dye and boild results in a lot of white precipitate. You can leave the TB in the sample if you want because the precipitate doesn't matter in SDS-PAGE as the gel below shows.
- Experiment that proves TB causes white precipitate:

100uL aliquots of the same overnight E. coli culture grown in TB medium were spun for ~3 minutes to pellet the cells. The TB media was left, replaced with water, or half was removed and replaced by water. Pellets were resuspended, and either home-made 4X Laemelli buffer or commercial Bio-Rad buffer was used. Cell suspensions were boiled for 5 minutes then photographed.
- Experiment that proves TB causes white precipitate:

Does that white precipitate after boiling matter?
- The picture above shows some frightening looking white precipitate. When these mixtures were run in a gel (after vortexing so lots of the white particulate was loaded), all samples ran the same:

white precipitate formed during boiling TB culture with 4X loading buffer doesn't affect the way the gel runs.

How much to load
Total Cell Protein
- use 5-8 or even up to 20 ug protein per well. (for Mini Protean with 10 well comb)
- Amanda runs 15 ug in skinny comb wells 2013/10/25
- 7.5 uL of turbid TB E. coli is about right for skinny wells. This becomes 10uL after adding 4X buffer.

sample of loading different uL*OD amounts of total BL21(DE3 culture. Protein = His*6 tagged ACS 2P2F

Purified Protein
- ~40 ng seems about right. -JM
- This means the minimum concentration is ~5ng/uL. When you load 7.5uL in the skinny-comb gels (becomes 10uL after adding 4X buffer) you get ~40 ng in the gel.
- 4 to 8 uL*uM works well for ~50-70kDa enzymes. If the stock is 1uM, add 4-8uL. If it is 4uM, add 1-2uL. *JM 20 August 2014

sample of loading different uL*uM amounts of protein. Protein = His*6 tagged ACS 2P2F

In general
- To avoid edge effects, add 1x sample buffer to unused wells. (from protein gel electrophoresis tips and troubleshooting guide)
- [[User:Janet B. Matsen|JM] doesn't do this 2014/3
- If biomass is not limiting, prep a significant excess of protein, which will allow you to re-run if your gel turns out poorly for any reason.
- Maximum volumes:
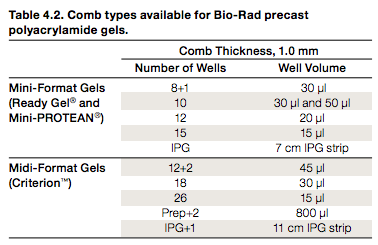
Maximum loading volumes for BioRad gels (source). As of 1/2014, the Lidstrom lab uses Mini-Format Gels with either 8 or 15 wells, so the maximum volumes are 30 or 15uL. Be careful not to let the samples splash into adjacent wells! - Load 12-15 uL, absolute max is 30 uL for 10-well comb in mini-protean
- Note: the amount you load affects the apparent size of the band:

The amount of protein loaded in an SDS-PAGE lane affects the apparent band size. Notice the difference between 6uL and 10 uL of the BL21(DE3) total protein lysates. The protein of interest is marked with a white dot/star
- Note: the amount you load affects the apparent size of the band:
- If gel is overloaded or under loaded, run a new gel with a different amount of dyed & boiled lysate


How to prepare samples
- If protein normalizing:
- lyse cultures by sonication. Use 20 1 second pulses while tube is in ice, let the samples rest 5 minutes on ice, then sonicate again. Wipe sonicator stick between uses. Don't touch the tip while it is on.
- sonication allows you access to un-dyed lysed sample, unlike boiling with sample loading buffer as is described below.
- do Pierce BCA BSA assay to determine concentration. Run each sample with dilutions: 1, 1/10, 1/20, etc.
- Boil samples with sample buffer + beta-mercaptoethanol
- Optional: check efficacy of lysis with light microscopy (people usually don't do this.)
- Centrifuge all extracts extensively (20,000 x g for 15 min at 15°C) to remove any insoluble mater
- You can run gels with the soluble, insoluble, and/or crude (not centrifuged) lysates.
- lyse cultures by sonication. Use 20 1 second pulses while tube is in ice, let the samples rest 5 minutes on ice, then sonicate again. Wipe sonicator stick between uses. Don't touch the tip while it is on.
- If not normalizing:
- Boil cultures with concentrated sample buffer + beta-mercaptoethanol
- need to prepare this mix from concentrated loading dye (pre-made) and beta-mercaptoethanol. Make this mix fresh each day.
- If using Amanda's mix, mix 10 uL of this mix (proportions below) into every 25 uL of cell culture.
- If using BioRad's mix, "dilute the samples at least 1:2 with sample buffer", which I presume is 1 part cell culture + 2 parts mix.
- Heat your sample by either: (link)
- a) Boiling for 5-10 minutes (Works for most proteins)
- b) 65 degrees C for 10 minutes (If you have smearing using the above procedure)
- c) 37 degrees for 30 minutes (Membrane proteins or others that do not enter the gel otherwise may benefit from this type of sample preparation)
- consider centrifugation to remove insoluble??
- heat sample to 37oC to solubilize precipitated SDS??
- Boil cultures with concentrated sample buffer + beta-mercaptoethanol
Consolidated/Pictorial Protocol

Running the Gel
- Bio-Rad Mini-Cell Setup
- If only 1 gel, use buffer dam to replace second gel
- Position the gels with the shorter plate facing inward!
- Apply pressure on gel holder and gels as you close the tabs to seal the center compartment.

- Fill central compartment with running buffer
- should fill sample wells
- Pour more into the outer compartment to specified line
- Load gel
- Make sure you will be able to determine the orientation of your gel after it is stained. Asymmetry is good!
- Make sure to color/charge-match the cords to the power unit as the electrodes in the gel holder to the contacts in the lid.
- Run @ 60 V for ~15 min, then 200 V for ~ 20+ min.
- Note: ladder looks blurry while running through the stacking gel; don't be alarmed unless it still looks blurry in the resolving gel.
- Can skip the 60V step if you don't need a gorgeous gel
- Amanda runs 20 min at 200V, then checks frequently to make sure the protein doesn't run off the gel.
- Lower voltages can be considered if 200V leads to poor resolution.
- "[the] best typical conditions for electrophoresis are at 10-15 V/cm." (reference: protein gel electrophoresis tips and troubleshooting guide)
Storing Samples After Use
- Amanda was taught to flash-freeze (liquid N2) samples and store them at -80oC. Janet loves liquid N2 (particularly dumping it on the ground after use) so I follow along without question.
- Ladder is stored at -80oC. An aliquot of PageRuler Prestained that sat out at room temp for 1 week looked perfect in a gel, so don't worry about that ladder's stability.
Staining, Destaining, & Visualization
Old school bromphenol blue dye
- dye overnight or cycles of 1 min @ power 6 in the microwave
- microwave by Bo's bench. Let it vent a little in the hood between heating events.
- Return dye to container
- Rinse to remove residual dye
- Destain (2 - 4 cycles)
- The destain we use is just as potent when diluted 1:1 with tap water. I do this to save material and destain solution mixing time. -JM 12/2012
- Save the destain from the 3rd & 4th cycles, and re-use it for a future gel's 1st destain.
- Image with white light transillumination
- use the white light box, set the Gel Logic program to "white light transillumination", and use an exposure time of about 0.15 seconds.
- Using 1 second (and a tighter aperture) results in a blurry image
- Using less than ~ 0.15 seconds leads to inconsistent exposures. (The final image won't usually match the exposure level shown in the preview screen.)
- use the white light box, set the Gel Logic program to "white light transillumination", and use an exposure time of about 0.15 seconds.

New school GelCode Blue Safe Protein Stain
- Product Info:
- GelCode Blue Safe Protein Stain: A coomassie gel stain that's safer and less costly to ship.
- Pierce Item #1860957, Item# 24594
- How to use:
- Wash gel 3x in tap water (says Frances). Let it marinate ~10 min each time.
- Pour enough on to cover gel
- stain 30 min - overnight. (Frances likes overnight)
- Dump leftover stain down drain
- you might consider re-using bugger if your bands are fat
- Destain in water, but not overnight (too long in Amanda's experience)
Other Resources
- Background (Wikipedia)
- Instructions for PageRuler protein standard
- Instructions for BioRad Rainbow Std
- Mini-PROTEAN® Tetra Cell manual
- more comprehensive BioRad manual for SDS-PAGE (Janet's favorite)
- Criterion manual (for larger sized gels)
- video how-to (not vetted)
- article about polymerization from BioRad
Mistakes to Be Careful About
Prepping Gels
- using an "old" APS solution when making gels. The 10% weight/volume APS can be made fresh each time for best results. Don't use a solution that is more than a month or so old. The gels won't polymerize.
- not mixing the liquid gel mixture enough
- Janet pipettes up and down with a 10 or 15 mL pipette about 10 times, being careful not to bubble the solution.
- letting the gel dry too long after pouring the stacking gel (comb step)
- the very edges can shrivel up, which becomes a problem when you try to use those edge lanes
- taking the gel off the casting stand before it has polymerized
- entire sample will leak out
- If using commercially prepared gels, don't forget to remove the tape on the bottom of the gel! If you load your samples and turn on the power and see voltage but no current, check for tape removal (duh, Nicole)!
Preparing Samples
- using too much beta-mercaptoethanol in your sample buffer
- should have < 1% beta-mercaptoethanol in the mix after you add sample buffer to the
- too much reduction of cysteines is bad: will alter structure and even cleave proteins.
Running Gels
- sample sloshing out of the well you are trying to load and falling into a neighboring well
- Electrode buffer leaking out from between the two plates
- Gel will run very unusually or stop running. Keep an eye out!
- avoid by making sure the plates are well-seated
- Using Tris-HCl instead of Tris in the electrode buffer
- not having the lid to the running unit on all the way. --> poor electrical contact & blurry bands
- Using resolving gel Tris Buffer instead of the intended electrode buffer.
- The results of said mistake: JM 2013/01/04

SDS-PAGE gel run with resolving gel buffer instead of the proper electrode buffer
- The results of said mistake: JM 2013/01/04

Visualizing Gels
- Gels tear easily when warm.
- Be sure not to tear the gel as you take it out. It is good to wet the glass after you remove the cover glass plate.
Recipes
All recipes except the staining & destaining solution are from the Mini-PROTEAN® Tetra Cell manual
10% (w/v) APS
- 0.10 g/1 mL diH2O = 100 mg/mL (prepare fresh daily for best results; most people do every few weeks.) (link)
Loading Buffer
- Mix pre-made concentrated loading buffer with fresh beta-mercaptoethanol prior to each use.
- TALON recipe: 5X SDS PAGE Sample Buffer
- 15% β-Mercaptoethanol (β-ME)
- 15% SDS
- 50% Glycerol
- 1.5% Bromophenol blue
- BioRad's loading buffer recipe:
- 3.55 mL deionized water, 1.25 mL 0.5 M Tris-HCl pH 6.8, 2.5 mL glycerol, 2.0 mL of 10% (w/v) SDS, 0.2 mL of 0.5% (w/v) Bromophenol Blue. Total volume = 9.5 mL.
- TALON recipe: 5X SDS PAGE Sample Buffer
- mix 50 uL beta-mercaptoethanol to 950 uL sample buffer prior to use.
- Scaled back 5x: 10 uL beta-mercaptoethanol + 190 uL 5x buffer
- scaled back 10x: 5 uL beta-mercaptoethanol + 95 uL buffer
- Dilute the sample "at least 1:2 with sample buffer" and heat at 95oC for 4 min to lyse the cells.
- Janet presumes this is 1 part sample per 2 parts buffer, but isn't sure (???)
- Amanda's recipe is a little different: 4x mix made of: 4 mL glycerol, 0.8 g SDS, 2.5 mL 1M Tris-HCl pH 6.8, 80 uL of bromophenol blue slurry (5 mg/mL = 0.5% (w/v)), H2O to 8 mL.
- Amanda mixes 160 uL 4X buffer, 8 uL beta-mercapto-ethanol, 32 uL H2O. Use 10 uL per 25 uL of cells. Recipe Comparison (to BioRad's)
- Can add up to 8M urea for really hydrophobic proteins
Running/Electrode Buffer:
- 10x SDS-PAGE (1 L) (BioRad catalog #161-0732) = 250 mM Tris, 1.92 M glycine, 1% SDS, pH 8.3
- Mix:
- Tris base 30.30 g DO NOT USE Tris-HCl
- Glycine 144.10 g
- SDS 10.00 g
- diH2O to 1 L
- Do not adjust the pH (~pH 8.3)
- Mix:
- Make 1L of 1x for use.
- Store at 4oC.
- This buffer is used while running proteins through the gel. Pour it in as the instructions for the box explain. Pour back into bottle for re-use afterward.
- It is best practice to not re-use electrode buffer. Our lab does, however, re-use the buffer ~ 4 (but sometimes up to 10) times.
Staining Buffer (Coomassie Brilliant Blue G-250):
- recipes vary. Usually there is about 0.5 g/L dye, and between 200 - 500 mL methanol per liter. 100 mL/L acetic acid is very common.
- 0.500 g Brilliant Blue, 500 mL methanol, 100 mL glacial acetic acid, 400 mL dH20. Mix well. Store at room temperature; can be reused 2-3 times.
- Amanda's recipe: 0.4 g of Coomassie Blue R 350 in 200 mL of 40% (v/v) methanol in water. Stir & filter (coffee filter is fine). Add 200 mL of 20% acetic acid in water (40 mL acetic acid in 160 mL water).
- note: Coomassie Brilliant Blue G-250 differs from Coomassie Brilliant Blue R-250 by the addition of two methyl groups. We use the G form. Read more about the R form here or at the bottom of this page.
Destaining Buffer:
- 30% methanol, 10% acetic acid, water
- some labs use much less methanol & acetic acid; some use plain water.
- Janet rinses in plain water before using our destaining buffer.
Acrylamide toxicity
- Acrylamide is toxic to your nervous system, and may be a carcinogen. The unpolymerized form is toxic, but the polymerized form is much less toxic. ALWAYS wear gloves and wipe up spills - once the solution dries, the dust can be inhaled. Interestingly, fried starchy/sugary foods naturally contain acrylamide, too.
- more than you want to know about acrylamide toxicity can be found here
Ladder
The Lidstrom Lab PageRuler ladder best. (manual)

Notes:
- The mobility of prestained proteins can vary in different SDS-PAGE buffer systems (manual)
- We load 4uL per well for 0.75mm thick gels; the manual suggests 5uL.
- You can't get use the ladders to approximate/quantify how much protein you added:
- "Thermo Scientific ladders are not designed for protein quantification. For quantification, we would recommend to use a protein of known concentration as a reference."
Other ladders:

Combs/Loading Volumes
- We currently only have the mini-protean gel running boxes. -JM 10/2012

Troubleshooting
- Hycult Biotech troubleshooting guide
- protein gel electrophoresis tips and troubleshooting guide includes troubleshooting after general tips
- nippongenetics guide
Questions/Answers/Facts
How do you prepare soluble and insoluble fractions for SDS-PAGE?
Yakov Kipnis of the Baker lab wrote in 11/2014: "Soluble and insoluble fractions of cell lysate look distinct on gel (ratios of band intensities for various proteins is quite different), therefore if you see large portion of unlysed cells contaminating insoluble fraction distinction between soluble/insoluble becomes less pronounced. To make the comparison easier I typically run 3 lanes/sample (total cell lysate/soluble(sup after spin)/insoluble(pellet after spin) I always try to load similar amount of protein on a gel. For "total" and "soluble" it is straightforward, to help solubilize as much of "insoluble" as possible I resuspend pellet in small volume of 8-9 M urea (avoid GuHCl it destroys SDS gels) first and reconstitute to the same volume before taking small aliquot for gel sample. Example: total volume of the lysate 500 uL (take 10 uL for gel="total"), spin (take 10 uL="soluble"), remove supernatant as much as possible, add 20-50 uL 9M urea, resuspend pellet, dilute to 490 uL by water/buffer (take 10 uL="insoluble"). This way theoretically overlay of "soluble" + "insoluble" lanes should give you "total", if corresponding bands do not sum up there is a chance something unaccounted happened. Later you can skip "total" to save space on gel."
What do all of the reagents do? How long do they last?
- SDS
- In loading buffer and often in gels (not necessary to include in gels; can be used in sample buffer)
- APS & TEMED
- polymerize acrylamide
- Stability:
- Biorad customer service 10/2012: "TEMED breaks down through interaction with oxygen, which produces many products in small amounts. TEMED, if stored properly, excluding oxygen, should not break down. It can pool up under the liner of the cap, and since it has very low viscosity, can creep between the threads of the cap and those of the bottle. TEMED color is a good indicator of degradation. TEMED becomes a darker yellow color when degraded. Our APS is guaranteed for 1 year at room temperature in dry form (powder). APS should last indefinitely if stored desiccated. "
- brand new BioRad TEMED opened 11/2012 was clear, not yellow. The old bottle was clear-yellow.
- glycine
- carry charge in the opposite direction as the negatively-charged SDS-covered proteins
- methanol
- in wash buffer
- glycerol in loading buffer: helps sample sink
- Bromphenol Blue
All about the "stacking" layer of the gel
- The stacking layer originally was a lower density acrylamide with a lower pH. The goal of "stacking" was to gather all the proteins into a tight band so that they all start from the "starting line" at the same time, and appear more clearly after completion. The lower pH of the stacking gel is intended to affect how much the glycine in the running buffer is attracted to the proteins, which affects the proteins mobility. This video explains it if you can tolerate the corniness.
- There are, however, different ways to achieve stacking:
- lower density gel on top that isn't at a different pH
- Example: commercially available gels from BioRad. The fixed-percentage gels have only one buffer ingredient.
- The fact that having different pH values in the stacking and resolving gels isn't so important clarifies why home-made gels that initially have different pHs in the two layers still work fine weeks after they were poured when presumably the buffers converge to equilibrium.
- a gradient of acrylamide percentages
- A stacking gel is not necessary when using a gradient gel, as the gradient itself performs this function. (thermo scientific)
- lower density gel on top that isn't at a different pH
Is it important to degas my water + buffer + acrylamide mix before adding the APS and TEMED, as the manuals recommend?
- "We recommend that you degas the solutions to get rid of nitrogen. Sometimes, the nitrogen may come out of solution and form bubbles in your gels. " -BioRad customer support 10/26/2012 JM
- "Proper degassing and filtering of the casting solution is critical for both reproducibility of the polymerization (oxygen removal)" (link)
- This manual suggests the consequence is poor polymerization. If you don't experience poor polymerization, maybe you don't need to worry about degassing...?
- Also note this is not the justification customer support provided. (see above)
Why shouldn't we overlay the gels with butanol or isopropanol as they polymerize?
- "We do not recommend using butanol/isopropanol because these may degrade the glue on the spacer plates and the plastic of the casting frame." -BioRad customer support 10/26/2012 JM
Should I soak my gel or run them without samples before use?
- They say no: "I have not heard that soaking the gels in water will improve the run. Talking with my colleagues, we thought it would have been detrimental because the tris and chloride ions will diffuse out during this time and in theory, should have made your gel run poorly."
- We had an episode in October & November 2012 where the gels ran funny unless they were pre-run without samples, but this was resolved by replacing gel-pouring buffers someone made with commercial BioRad buffers. About the temporary issue:
- Ladder (and all other protein samples) ran fine until 1/2 way through the gel, after which they nearly halted and stacked on top of one another. Immediately after, a ~1 cm wide band that spanned across the gel was visible as it refracted light differently. This band disappeared after a while of soaking, leading me to believe it was a buildup of one of the buffer compounds. When such a gel was soaked overnight in water and ladder was loaded in an unused adjacent well, and it ran perfectly without bunching up. This was a very repeatable phenomenon with all gels used before the problem was solved.
- Potential problems with soaking or pre-running gels:
- You might ruin the stacking nature of the gel by altering the buffer within the acrylamide matrix. This essentially converts your gel to a continuous buffer gel system, known to give more blurry bands.
Is it ok to re-use my electrode buffer?
- Many people re-use it up to ~ 20 times, however, you should know that the manuals recommend single use and understand why. This may help.
How long can I store my acrylamide gels?
- "Tris-HCl resolving gels are prepared at pH 8.6–8.8. At this basic pH, polyacrylamide slowly hydrolyzes to polyacrylic acid, which can compromise separation. For this reason, Tris-HCl gels have a relatively short shelf life. In addition, the gel pH can rise to pH 9.5 during a run, causing proteins to undergo deamination and alkylation. This may diminish resolution and complicate post electrophoresis analysis." (reference)
- note: commercial Tris gels have a shelf life of 6 months to 1 year. Amanda & Ceci don't keep hand-poured gels for more than a few (~4) weeks. (JM 11/2012)
- JM uses gels that are up to 11 weeks old. They work perfectly. I haven't tested longer yet.
- It is possible the commercial gels have other means of extending shelf life.
- note: commercial Tris gels have a shelf life of 6 months to 1 year. Amanda & Ceci don't keep hand-poured gels for more than a few (~4) weeks. (JM 11/2012)
What is the difference between Coomassie R and Coomassie G?
- useful link
- note: Coomassie Brilliant Blue G-250 differs from Coomassie Brilliant Blue R-250 by the addition of two methyl groups. We use the G form. Read more about the R form here.
- R in R-250 stands for Reddish hue while G in G-250 for Greenish hue. R-250 is dark reddish blue/purple stain while G-250 gives lighter greenish blue stain.
BioRad once told Amanda:
- The G-250 form the colloidal particles in an aqueous solution. This is an advantage for staining a gel because the colloids tend not to stain the gel matrices, reducing the background problem. When the colloids come close to the proteins, the dye molecule is removed from the colloids by the nearby proteins due to the higher affinity of proteins to the dye.
- R-250, on the other hand, doesn't form the colloids. Rather, an individual dye molecule is dispersed in a solution. Therefore, the dye molecules can interact not only with proteins but with gel matrices freely, creating the background staining issue.
Should I throw my gel away if I forgot to add SDS to the resolving gel?
- Janet thinks no, for two reasons:
- The manual says "Bio-Rad’s precast gels also do not contain SDS and can be used for native or denaturing PAGE." (citation)
- In 11/2012 I forgot to, and the proteins ran wonderfully, anyway.
Other Tips
Collecting samples in PCR strip tubes makes life nice!
- You can concentrate samples using the PCR strip spinner on Marina's bench. It must be balanced!
- You can do boiling-driven lysis in a thermocycler
- Samples don't take up a lot of room in the freezer.
- To write on them (for recording the date, and strip number if you have multiple) you can use a fine-tip sharpie and cover it with a ring of scotch-tape. Without tape, the writing tends to wear off.
- We are in love with individually removable caps that stay attached as is sold here.
- Samples don't splattering when you take of the caps, reducing cross-contamination and mess
- If you only have one cap open at a time when loading your gel, you can tell much more easily which sample you just loaded, reducing the possibility that you have a problem.
- Do, still, take each cap off slowly when opening them if there is liquid in the lid of the chamber. Often after shaking the strip to mix it, I open the cap quickly and splurt the blue sample on my clothes. It always rinses clean after adding water, but it is nicer if this doesn't happen.
Excess salt in SDS-PAGE samples causes fuzzy bands and narrowing of gel lanes toward the bottom of the gel
- If the ionic strength is very high, no bands will appear in the lower part of the gel (a vertical streak will appear instead) and the dye front will be wavy instead of straight. Deionize any sample with a total ionic strength over 50 mM using columns such as Micro Bio-Spin™ columns, which contain 10 mM Tris at a pH suitable for SDS-PAGE. (source)
Success or failure of any protein analysis depends on sample purity.
(source)
- Interfering substances that can negatively impact SDS-PAGE include salts, detergents, denaturants, or organic solvents (Evans et al. 2009). Highly viscous samples indicate high DNA and/or carbohydrate content, which may also interfere with PAGE separations. In addition, solutions at extreme pH values (for example, fractions from ion exchange chromatography) diminish the separation power of most electrophoresis techniques. Use one of the following methods as needed to remove these contaminants:
- Protein precipitation — the most versatile method to selectively separate proteins from other contaminants consists of protein precipitation by trichloroacetic acid (TCA)/acetone followed by resolubilization in electrophoresis sample buffer. A variety of commercial kits can simplify and standardize laboratory procedures for protein isolation from biological samples
- Buffer exchange — size exclusion chromatography is another effective method for removing salts, detergents, and other contaminants
Polyacrylamide is oxidative, and so disulfide bonds may be re-formed after proteins enter the gel
- also, oxidation of agents used to reduce any disulfide bonds in the original sample may introduce artifactual new components in the gel pattern. (source)
SDS can form micelles
- The SDS concentration is greater than the critical micelle concentration (cmc), so SDS is present as micelles. Micellar SDS bound to the tracking dye unstacks at high gel concentrations in disc electrophoresis; so a tracking dye that marks the moving boundary front at low gel concentrations fails to do so at high concentrations (source)
KCl causes SDS to precipitate
- If you samples contain KCl you should dilute them or methanol precipitate them and resuspend them in 1X sample buffer. With low concentrations of KCl (<200 mM) you can run them on the gel but you should load every lane with sample buffer containing the same concentration of KCl (even if they are blanks). This will help the gel run a little less anomalously. (source)
Supplies
- BioRad sells most of the reagents we use. You can make most of them yourself, if you'd like.
- The "diaper" that goes under the gel casting stand:
- Janet called BioRad , UW EH&S, and VWR on 11/29/2012 to identify a special pad that is particularly good for absorbing liquid acrylamide mixtures used in pouring acrylamide gels. The idea is that such a pad would prevent the dried acrylamide from being re-released into the air and breathed by scientists. Unfortunately, this doesn't seem to exist.
- We ordered these