*When the PCR finished, I ran a small [[/20060802Sections56Fusion|gel]] containing 3 lanes for each of the 3 tubes.
*When the PCR finished, I ran a small [[Media:20060802Sections56Fusion.jpg|gel]] containing 3 lanes for each of the 3 tubes[[Image:20060802Sections56Fusion.jpg|thumb|right|2 part assembly with sections 5 and 6]].
*I made more 1% agarose:
*I made more 1% agarose:
**5g Agarose in 500mL of TAE, microwaved for 4 minutes at 100% power.
**5g Agarose in 500mL of TAE, microwaved for 4 minutes at 100% power.
Line 1,072:
Line 1,072:
**#72.0*C for 10 minutes
**#72.0*C for 10 minutes
**#4*C for 1 minute
**#4*C for 1 minute
*When the PCR finished, I ran a small [[/20060803Sections567fusiontrial1|gel]] at 40V.
*When the PCR finished, I ran a small [[Media:20060803Sections567fusiontrial1.jpg|gel]] at 40V[[Image:20060803Sections567fusiontrial1.jpg|thumb|right|gel results of the first attempt at making a 3 part assembly (sections 5 through 7)]].
**I ran 3 lanes of 5-7A, 3 lanes of 5-7B, and 3 lanes of 5-7C.
**I ran 3 lanes of 5-7A, 3 lanes of 5-7B, and 3 lanes of 5-7C.
**The PCR did not work.
**The PCR did not work.
Line 1,100:
Line 1,100:
**#4*C for 1 minute
**#4*C for 1 minute
***I increased the extension times because in the previous PCR program, the extension times were not consistent with yesterday's PCR, which worked.
***I increased the extension times because in the previous PCR program, the extension times were not consistent with yesterday's PCR, which worked.
*During this PCR, I ran another [[/20060803Sections567fusiontrial1gel2|gel]]:
*During this PCR, I ran another [[Media:20060803Sections567fusiontrial1gel2.jpg|gel]][[Image:20060803Sections567fusiontrial1gel2.jpg|thumb|right|second gel image of the 3 part fusion attempt]]:
**Lanes 1-2: 5-7A
**Lanes 1-2: 5-7A
**Lanes 3-4: 5-7B
**Lanes 3-4: 5-7B
Line 1,110:
Line 1,110:
**There are faint bands below the 100bp marker.
**There are faint bands below the 100bp marker.
*When the new PCR finished, I placed the tubes in the 4*C fridge.
*When the new PCR finished, I placed the tubes in the 4*C fridge.
*I ran a small [[/20060803Sections567FusionTrial2|gel]] on the afternoon's PCR:
*I ran a small [[Media:20060803Sections567FusionTrial2.jpg|gel]] on the afternoon's PCR[[Image:20060803Sections567FusionTrial2.jpg|thumb|right|2nd attempt at a 3 part assembly (with sections 5 through 7)]]:
**Lanes 1-2: 5-7D
**Lanes 1-2: 5-7D
**Lanes 3-4: 5-7E
**Lanes 3-4: 5-7E
Line 1,186:
Line 1,186:
====August 7,2006====
====August 7,2006====
*I ran a small [[/20060807Sections567FusionTrial3|gel]] at 120V.
*I ran a small [[Media:20060807Sections567FusionTrial3.jpg|gel]] at 120V[[Image:20060807Sections567FusionTrial3.jpg|thumb|right|successful 3 part fusion (sections 5 through 7)]].
**Lane 1: 2-log ladder
**Lane 1: 2-log ladder
**Lanes 2-3: 5-7G
**Lanes 2-3: 5-7G
Line 1,251:
Line 1,251:
**#72.0*C for 10 minutes
**#72.0*C for 10 minutes
**#4*C for 1 minute
**#4*C for 1 minute
*When this PCR finished running, I ran a small [[/20060807Sections5678FusionTrial1|gel]] at 120V.
*When this PCR finished running, I ran a small [[Media:20060807Sections5678FusionTrial1.jpg|gel]] at 120V[[Image:20060807Sections5678FusionTrial1.jpg|thumb|right|sections 5 through 8 assembly product]].
**Lane 1: 2-log ladder
**Lane 1: 2-log ladder
**Lanes 2-3: 5-8A
**Lanes 2-3: 5-8A
Line 1,378:
Line 1,378:
**#72.0*C for 10 minutes
**#72.0*C for 10 minutes
**#4*C for 1 minute
**#4*C for 1 minute
*When the PCR finished, I ran a [[/20060808Sections1Through9FusionTrial1|gel]] at 120V.
*When the PCR finished, I ran a [[Media:20060808Sections1Through9FusionTrial1.jpg|gel]] at 120V.
[[Image:20060808Sections1Through9FusionTrial1.jpg|thumb|right|attempted 9 part assembly (section 1 through 9)]]
**Lane 1: 2-log ladder
**Lane 1: 2-log ladder
**Lanes 2-3: 1-9A
**Lanes 2-3: 1-9A
Line 1,387:
Line 1,388:
**There is no band at the 8520bp position (expected product of the fusion of sections 1 through 9) in any lane. There is a light band between the 5kb and 6kb position in lanes 2 and 3. Tomorrow I will run a gel on all of my gel extracts to confirm that my template stocks are okay.
**There is no band at the 8520bp position (expected product of the fusion of sections 1 through 9) in any lane. There is a light band between the 5kb and 6kb position in lanes 2 and 3. Tomorrow I will run a gel on all of my gel extracts to confirm that my template stocks are okay.
**There is smearing in the 2-log ladder, but not in the 1kb ladder. This suggests that there may be something wrong with the 2-log ladder stock.
**There is smearing in the 2-log ladder, but not in the 1kb ladder. This suggests that there may be something wrong with the 2-log ladder stock.
====August 9, 2006====
*I ran a PCR to attempt to fuse sections 5, 6, 7, 8, and 9:
**Master Mix:
***148uL nuclease-free water
***40uL phusion hf buffer
***2uL 25mM dNTPs
***2uL section 5 template
***2uL section 6 template
**I added 48.5uL of master mix into each of 3 tubes.
**I labeled these tubes 5-9A, 5-9B, 5-9C.
**I added 0.5uL phusion DNA polymerase to each tube.
**I ran the following short program with the PCR machine:
**#98.0*C for 30 seconds
**#98.0*C for 10 seconds
**#55.0*C for 30 seconds
**#72.0*C for 30 seconds
**#go to 2, 1 time
**#4*C for 1 minute
**I added 0.5uL of section 7 template (from 7A) to 5-9A
***New volume: 49.5uL
**I added 1uL of section 7 template (from 7A) to 5-9B
***New volume: 50uL
**I added 1.5uL of section 7 template (from 7A) to 5-9C
***New volume: 50.5uL
**I ran the following short program on the 3 tubes:
**#98.0*C for 30 seconds
**#98.0*C for 10 seconds
**#60.0*C for 30 seconds
**#72.0*C for 50 seconds
**#go to 2, 1 time
**#4*C for 1 minute
**I added 0.5uL of section 8 template to 5-9A
***New volume: 50uL
**I added 1uL of section 8 template to 5-9B
***New volume: 51uL
**I added 1.5uL of section 8 template to 5-9C
***New volume: 52uL
**I ran the following short program on the 3 tubes:
**#98.0*C for 30 seconds
**#98.0*C for 10 seconds
**#60.0*C for 30 seconds
**#72.0*C for 1 minute 10 seconds
**#go to 2, 1 time
**#4*C for 1 minute
**I added 0.5uL of section 9 template (from 9A) to 5-9A
***New volume: 50.5uL
**I added 1uL of section 9 template (from 9A) to 5-9B
***New volume: 52uL
**I added 1.5uL of section 9 template (from 9A) to 5-9C
***New volume: 53.5uL
**I ran the following short program on the 3 tubes:
**#98.0*C for 30 seconds
**#98.0*C for 10 seconds
**#60.0*C for 30 seconds
**#72.0*C for 1 minute 30 seconds
**#go to 2, 1 time
**#4*C for 1 minute
**I added 0.5uL of section 5 forward primer to each tube and 0.5uL of section 9 reverse primer to each tube.
***New volumes:
****5-8A: 51.5uL
****5-8B: 53uL
****5-8C: 54.5uL
**I ran the following long PCR program:
**#98.0*C for 30 seconds
**#98.0*C for 10 seconds
**#65.0*C for 30 seconds
**#72.0*C for 1 minute 50 seconds
**#go to 2, 35 times
**#72.0*C for 10 minutes
**#4*C for 1 minute
*When the PCR finished, I ran a [[Media:20060809Sections56789FusionTrial1.jpg|gel]] at 120V.
[[Image:20060809Sections56789FusionTrial1.jpg|thumb|right|gel showing the 5 part assembly (sections 5 through 9)]]
**Lane 1: 2-log ladder
**Lanes 2-3: 5-9A
**Lanes 4-5: 5-9B
**Lanes 6-7: 5-9C
**Lanes 8-9: 9A
**Lane 10: 1kb ladder
**Samples 5-9B and 5-9C had stronger bands at the 5239bp position (expected length of sections 5 through 9 combined). This suggests that it is better to add more template between PCR steps.
*Experimental factors to consider:
**Ratio of templates: to increase our chances for the intermediate PCR steps to create the combined template, we should try adding increasing amounts of DNA template between PCR steps.
**Ratio of the reaction mixture: since the reaction volume changes throughout the process, we may need to experiment with increased amounts of buffer, dNTPs, DNA polymerase, and primers.
**Rounds of PCR: we will need to see what the optimal number of rounds is for each intermediate short PCR to more efficiently or quickly get the combined sections template.
**Annealing temperature and extension time per additional template: I have been adding a standard 20 seconds to the extension time each time a template is added. This may be too much for certain sections.
**Buffer: we should try using GC buffer, which is supposedly better for long DNA sequences.
*I realize that in adding up the combined lengths of my sequences, I forgot to take into account the 30bp overlaps between each of the 9 T7+ alpha sections. My calculated lengths should actually be a bit shorter - I will recalculate these values later.
====August 16, 2006====
*I ran a PCR to amplify the sections 5-9 fusion pcr from last week.
*I ran a [[Media:20060816CombinedSections5Through9Amplification.jpg|gel]] on the PCR products.
[[Image:20060816CombinedSections5Through9Amplification.jpg|thumb|right|gel image of the attempted amplification of sections 5 through 9 from the sections 5 through 9 assembly product]]
**Lane 1: 2-log ladder
**Lanes 2-3: 5-9 PCR 1
**Lanes 4-5: 5-9 PCR 2
**Lanes 6-7: 5-9 PCR 3
**Lanes 8-9: 5-9C
**Lane 10: 1kb ladder
**The amplifications of 5-9C did not work. There are also many other bands in the PCR. We will figure out what those bands are later. Tomorrow I will run a gel on 5-9C and 5-9B to gel isolate the sections 5-9 combined template.
*I ran a PCR to amplify the sequence from section 2 through section 9 on the T7+ DNA. This will test the range of phusion DNA polymerase.
**Master Mix:
***120.25uL nuclease-free water
***32.5uL 5x phusion hf buffer
***1.625uL 25mM dNTPs
***3.25uL T7+ DNA
***1.625uL section 2 forward primer
***1.625uL section 9 reverse primer
**I added 49.5uL of MM to each of three tubes labeled 2-9A, 2-9B, and 2-9C.
**I added 0.5uL of phusion DNA polymerase to each tube.
**I ran the following program on a gradient block:
**#98.0*C for 30 seconds
**#98.0*C for 10 seconds
**#55.0*C (column 1, 2-9A), 59.3*C (column 6, 2-9B), and 65.0*C (column 12, 2-9C) for 30 seconds
**#72.0*C for 2 minutes 20 seconds
**#go to 2, 35 times
**#72.0*C for 10 minutes
**#4*C for 1 minute
*I ran a [[Media:20060816AllAlphaSectionsGel1.jpg|gel]] to test the quality of the template for each section.
[[Image:20060816AllAlphaSectionsGel1.jpg|thumb|right|gel image of each section's template]]
**Lane 1: 2-log ladder
**Lane 2: Section 1 Template (from 1A)
**Lane 3: Section 2 Template
**Lane 4: Section 3 Template (from 3A)
**Lane 5: Section 4 Template (from 4A)
**Lane 6: Section 5 Template
**Lane 7: Section 6 Template
**Lane 8: Section 7 Template (from 7A)
**Lane 9: Section 8 Template
**Lane 10: Section 9 Template (from 9A)
**The lanes were clean and had bands in all the correct positions.
*We will need to make more of sections 6 and 8 template.
====August 17, 2006====
*I ran a large [[Media:20060817Sections5Through9FusionGelExtraction.jpg|gel]] on 5-9B and 5-9C.
[[Image:20060817Sections5Through9FusionGelExtraction.jpg|thumb|right|gel isolation of the sections 5 through 9 assembly product]]
**Lane 1: 2-log ladder
**Lanes 2-4: 5-9B
**Lanes 5-6: 5-9C
*While the gel ran, I ran a PCR assembly on sections 1, 2, and 3.
**I made a master mix:
***129.5uL nuclease-free water
***35uL 5x phusion hf buffer
***1.75uL 25mM dNTPs
***1.75uL section 1 template (from 1A)
***1.75uL section 2 template
**I may have accidentally added 2uL each of the dNTPs and the templates, instead of 1.75uL.
**I added 48.5uL of MM into each of three tubes: 1-3A, 1-3B, and 1-3C.
**I added 0.5uL of phusion DNA polymerase to each tube for a final volume of 49uL in each tube.
**I ran the following extension program on the three tubes:
**#98.0*C for 30 seconds
**#98.0*C for 10 seconds
**#55.0*C for 30 seconds
**#72.0*C for 30 seconds
**#go to 2, 1 time
**#4*C for 1 minute
**I added 0.5uL of section 3 template (from 3A) to 1-3A, 1uL to 1-3B, and 1.5uL to 1-3C.
**New volumes: 1-3A: 49.5uL, 1-3B: 50uL, and 1-3C: 50.5uL.
**I ran another extension program on the tubes:
**#98.0*C for 30 seconds
**#98.0*C for 10 seconds
**#60.0*C for 30 seconds
**#72.0*C for 40 seconds
**#go to 2, 1 time
**#4*C for 1 minute
**I added 1uL of section 1 forward primer and 0.5uL of section 3 reverse primer to each tube.
**Final volumes: 1-3A: 51uL, 1-3B: 51.5uL, and 1-3C: 52uL.
**I ran a final PCR step to amplify the combined sections 1 - 3.
**#98.0*C for 30 seconds
**#98.0*C for 10 seconds
**#65.0*C for 30 seconds
**#72.0*C for 1 minute
**#go to 2, 35 times
**#72.0*C for 10 minutes
**#4*C for 1 minute
*When my gel of the section 5-9 fusion finished running, I used the Qiagen Gel Extraction kit to purify the combined sections 5-9 band.
**5-9B: 350uL gel volume
***30uL final volume at 6.4ng/uL
**5-9C: 250uL gel volume
***30uL final volume at 5.7ng/uL
*I ran a [[Media:20060817Sections2Through9Amplification.jpg|gel]] on yesterday's PCR product of the sections 2 through 9 amplification.
[[Image:20060817Sections2Through9Amplification.jpg|thumb|right|gel results of the amplification of sections 2 through 9 off of T7 wildtype DNA]]
**Lane 1: 2-log ladder
**Lanes 2-4: 2-9A
**Lanes 5-7: 2-9B
**Lanes 8-10: 2-9C
**I observed no bands at the 7943bp position (expected length). The PCR of long sequences needs further optimization.
*I ran a [[Media:20060817Sections123PCRAssemblyTrial1.jpg|gel]] on the PCR assembly of sections 1, 2, and 3 from earlier today.
[[Image:20060817Sections123PCRAssemblyTrial1.jpg|thumb|right|gel results of the PCR assembly of sections 1 through 3]]
**The gel showed no bands at the 3091bp position (expected length of assembly). The extension times for each assembly step may not be long enough. I may need to design a new procedure to accomodate the small sizes of sections 1 and 3 (367bp and 130bp).
====August 18, 2006====
*I ran a PCR assembly to attempt to put together sections 1 and 2.
**Master Mix:
***129.5uL nuclease-free water
***35uL 5x phusion hf buffer
***1.75uL 25mM dNTPs
***1.75uL section 1 template
***1.75uL section 2 template
**I added 48.5uL of MM to each of three tubes: 1-2A, 1-2B, and 1-2C.
**I added 0.5uL phusion DNA polymerase
**I ran the following short assembly program:
**#98.0*C for 30 seconds
**#98.0*C for 10 seconds
**#55.0*C for 30 seconds
**#72.0*C for 30 seconds
**#go to 2, 1 time
**#4*C for 1 minute
**I added 1uL of section 1 forward primer and 0.5uL of section 2 reverse primer to each of the three tubes.
**Final volumes: 50.5uL
**I ran the following PCR program:
**#98.0*C for 30 seconds
**#98.0*C for 10 seconds
**#65.0*C for 30 seconds
**#72.0*C for 40 seconds
**#go to 2, 35 times
**#72.0*C for 10 minutes
**#4*C for 1 minute
*Sri will take out my PCR at 3:00PM.
====August 21, 2006====
*I ran a small [[Media:20060821Sections12AssemblyTrial1.jpg|gel]] on the results of the sections 1 and 2 assembly from Friday. [[Image:20060821Sections12AssemblyTrial1.jpg|thumb|right|gel of results of the sections 1 and 2 assembly]]
**Lane 1: 2-log ladder
**Lanes 2-3: 1-2A
**Lanes 4-5: 1-2B
**Lanes 6-7: 1-2C
**Lane 8: Section 1 Template (from 1A)
**Lane 9: Section 2 Template
**Lane 10: 2-log ladder
**The gel showed bands around the 750bp position. Our expected length for the section 1 plus section 2 assembly was 2041bp.
*I PCR amplified my sections 5 through 9 assembly from August 9, 2006. I will use my gel extract from August 17, 2006.
**I made a Master Mix:
***259uL nuclease-free water
***70uL 5x phusion hf buffer
***3.5uL 25mM dNTPs
***3.5uL section 9 reverse primer
**I put 48uL of MM into each of 6 tubes.
**I put 1uL of T7+ DNA into each of 3 tubes labeled 2-9D, 2-9E, and 2-9F.
**I put 0.5uL of section 2 forward primer into each of these tubes.
**In the other 3 tubes, I put 1uL of assembled sections 5-9 and labeled these tubes 5-9 AA, 5-9 AB, 5-9 AC.
**I put 0.5uL of section 5 reverse primer into each of these 3 tubes.
**I put 0.5uL of phusion DNA polymerase into each of the 6 tubes.
**I ran the following PCR program:
**#98.0*C for 30 seconds
**#98.0*C for 10 seconds
**#65.0*C for 30 seconds
**#72.0*C for 2 minutes (2-9D, 5-9AA), 3 minutes (2-9E, 5-9AB), and 4 minutes (2-9F, 5-9AC)
**#go to 2, 35 times
**#72.0*C for 10 minutes
**#4*C for 1 minute
**These PCRs will run overnight.
*I should test a section 4 + 5 assembly as a comparison to the section 1 + 2 assembly. I think there may be a problem with trying to fuse two DNA segments that are very different in size.
*The sections 1 + 2 + 3 assembly and the sections 1 through 9 assembly may not have worked because of:
**the size differences between sections 1 and 2, and 4 and 5
**unspecific binding by the section 3 reverse primer
**unspecific binding by the section 1 forward primer
*I should also test the an assembly with sections 2 + 3 and sections 4 + 5 because on my very first attempt at PCR amplification of the sections from the T7+ DNA, all these sections had a second PCR product which was not the correct size.
*I may want to try using more assembly rounds for the sections 1 + 2 and 4 + 5 assemblies.
====August 22, 2006====
*I ran a PCR assembly on sections 4 and 5.
**Master Mix:
***129.5uL nuclease-free water
***35uL 5x phusion hf buffer
***1.75uL 25mM dNTPs
***1.75uL section 4 template (from 4A)
***1.75uL section 5 template
**I added 48.5uL of MM to each of three tubes: 4-5A, 4-5B, and 4-5C.
**I added 0.5uL phusion DNA polymerase
**I ran the following short assembly:
**#98.0*C for 30 seconds
**#98.0*C for 10 seconds
**#50.0*C for 30 seconds
**#72.0*C for 30 seconds
**#go to 2, 1 time
**#4*C for 1 minute
**I added 0.5uL of section 4 forward primer and 0.5uL of section 5 reverse primer to each of the three tubes.
**Final volumes: 50uL
**I ran the following PCR program:
**#98.0*C for 30 seconds
**#98.0*C for 10 seconds
**#65.0*C for 30 seconds
**#72.0*C for 30 seconds
**#go to 2, 35 times
**#72.0*C for 10 minutes
**#4*C for 1 minute
*During the PCR, I ran a small [[Media:20060822Sections2Through9AmplificationTrial2.jpg|gel]] on yesterday's PCR amplification of sections 2 through 9 from the T7+ DNA.
[[Image:20060822Sections2Through9AmplificationTrial2.jpg|thumb|right|gel results of yesterday's PCR amplficication of sections 2 through 9 from the T7 wildtype DNA]]
**Lane 1: 2-log ladder
**Lanes 2-4: 2-9A
**Lanes 5-7: 2-9B
**Lanes 8-10: 2-9C
**The gel did not show any bands above 3kb.
*When the first PCR finished, I ran a new PCR assembly of sections 2 and 3.
**Master Mix:
***129.5uL nuclease-free water
***35uL 5x phusion hf buffer
***1.75uL 25mM dNTPs
***1.75uL section 2 template
***1.75uL section 3 template (from 3A)
**I added 48.5uL of MM to each of three tubes: 2-3A, 2-3B, and 2-3C.
**I added 0.5uL phusion DNA polymerase
**I ran the following short assembly:
**#98.0*C for 30 seconds
**#98.0*C for 10 seconds
**#50.0*C for 30 seconds
**#72.0*C for 40 seconds
**#go to 2, 1 time
**#4*C for 1 minute
**I added 0.5uL of section 2 forward primer and 0.5uL of section 3 reverse primer to each of the three tubes.
**Final volumes: 50uL
**I ran the following PCR program:
**#98.0*C for 30 seconds
**#98.0*C for 10 seconds
**#65.0*C for 30 seconds
**#72.0*C for 1 minute
**#go to 2, 35 times
**#72.0*C for 10 minutes
**#4*C for 1 minute
*During this PCR, I ran a small [[Media:20060822CombinedSections5Through9AmplificationTrial2.jpg|gel]] on yesterday's PCR amplification of sections 2 through 9 from the T7+ DNA[[Image:20060822CombinedSections5Through9AmplificationTrial2.jpg|thumb|right|gel results of yesterday's PCR amplficication of the section 5 through 9 assembly gel extract]].
**Lane 1: 2-log ladder
**Lanes 2-3: 5-9AA
**Lanes 4-5: 5-9AB
**Lanes 6-7: 5-9AC
**Lanes 8-9: 5-9B
**Lane 10: 2-log ladder
**Lanes 2-7 showed no bands at the position of the band from lanes 8 and 9. Tomorrow I will try amplifying the section 5 through 8 assembly. I will also try amplifying sections 5-6, 5-7, 5-8, and 5-9 from wildtype DNA.
====August 23, 2006====
*I ran a PCR assembly on sections 3 and 4.
**Master Mix:
***129.5uL nuclease-free water
***35uL 5x phusion hf buffer
***1.75uL 25mM dNTPs
***1.75uL section 3 template (from 3A)
***1.75uL section 4 template (from 4A)
**I added 48.5uL of MM to each of three tubes: 3-4A, 3-4B, and 3-4C.
**I added 0.5uL phusion DNA polymerase
**I ran the following short assembly:
**#98.0*C for 30 seconds
**#98.0*C for 10 seconds
**#65.0*C for 30 seconds
**#72.0*C for 20 seconds
**#go to 2, 1 time
**#4*C for 1 minute
**I added 0.5uL of section 3 forward primer and 0.5uL of section 4 reverse primer to each of the three tubes.
**Final volumes: 50uL
**I ran the following PCR program:
**#98.0*C for 30 seconds
**#98.0*C for 10 seconds
**#65.0*C for 30 seconds
**#72.0*C for 20 seconds
**#go to 2, 35 times
**#72.0*C for 10 minutes
**#4*C for 1 minute
*I ran a large [[Media:20060823Sections5Through8FusionGelExtraction1.jpg|gel]] on my PCR assembly product of sections 5 through 8[[Image:20060823Sections5Through8FusionGelExtraction1.jpg|thumb|right|gel isolation of the assembly product of the sections 5 through 8 pcr fusion]].
**Lane 1: 2-log ladder
**Lanes 2-3: 5-8C
*I used the Qiagen Gel Extraction Kit to isolate the sections 5 though 8 assembly product.
**5-8ASS 230uL gel volume
***30uL final volume at 16.4ng/uL
====August 24, 2006====
*I ran a PCR to amplify sections 5 through 8 off of the sections 5 through 8 4 part assembly.
**Master Mix:
***259u nuclease-free water
***70uL 5x phusion hf buffer
***3.5uL 25mM dNTPs
***3.5uL section 5 forward primer
***3.5uL section 8 reverse primer
**I put 48.5uL into each of 6 tubes labeled 5-8AA, 5-8AB, 5-8AC, 5-8 T7+ A, 5-8 T7+ B, and 5-8 T7+ C.
**I put 1uL of the gel extracted sections 5 through 8 assembly product into each of tubes 5-8AA, 5-8AB, and 5-8AC.
**I put 1uL of T7+ DNA into each of tubes 5-8 T7+ A, 5-8 T7+ B, and 5-8 T7+ C.
**I added 0.5uL of phusion DNA polymerase to each of the six tubes.
**I ran the following PCR program:
**#98.0*C for 30 seconds
**#98.0*C for 10 seconds
**#65.0*C for 30 seconds
**#72.0*C for 2 minutes (5-8AA and 5-8 T7+ A), 3 minutes (5-8AB and 5-8 T7+ B), and 4 minutes (5-8AC and 5-8 T7+ C).
**#go to 2, 35 times
**#72.0*C for 10 minutes
**#4*C for 1 minute
*I ran a small [[Media:20060824Sections4And5AssemblyTrial1.jpg|gel]] on the sections 4 and 5 assembly from August 22, 2006[[Image:20060824Sections4And5AssemblyTrial1.jpg|thumb|right|pcr assembly of sections 4 and 5]].
**Lane 1: 2-log ladder
**Lanes 2-3: 4-5A
**Lanes 4-5: 4-5B
**Lanes 6-7: 4-5C
**Lane 8: Section 4 template (from 4A)
**Lane 9: Section 5 template
**Lane 10: 2-log ladder
**It appears that the assembly may have worked, but it is too hard to tell, since the expected product is not much bigger than section 5 itself. I will have to run this gel again, and for longer, in order to look for any separation between lanes 2 through 7 and lane 9.
*I ran a small [[Media:20060824Sections2And3AssemblyTrial1.jpg|gel]] on the sections 2 and 3 assembly from August 22, 2006[[Image:20060824Sections2And3AssemblyTrial1.jpg|thumb|right|pcr assembly of sections 2 and 3]].
**Lane 1: 2-log ladder
**Lanes 2-3: 2-3A
**Lanes 4-5: 2-3B
**Lanes 6-7: 2-3C
**Lane 8: Section 2 template
**Lane 9: Section 3 template (from 3A)
**Lane 10: 2-log ladder
**The expected product of the assembly is 2754bp. There is a strong band in each of lanes 2-7 that is near the 3kb position, which indicates that this assembly worked.
====August 28. 2006====
*I ran a small [[Media:20060825Sections3And4AssemblyTrial1.jpg|gel]] on the pcr assembly of sections 3 and 4 from August 23, 2006[[Image:20060825Sections3And4AssemblyTrial1.jpg|thumb|right|gel results from a pcr assembly of sections 3 and 4]].
**Lane 1: 2-log ladder
**Lanes 2-3: 3-4A
**Lanes 4-5: 3-4B
**Lanes 6-7: 3-4C
**Lane 8: Section 3 template (from 3A)
**Lane 9: Section 4 template (from 4A)
**Lane 10: 2-log ladder
**It is difficult to tell the assembly bands apart from the section 3 template band, with respect to size. I will have to run this gel again, but longer.
*I ran a small [[Media:20060825CombinedSections5Through8AmplificationTrial1.jpg|gel]] on yesterday's pcr amplification of the gel isolated pcr assembly product of sections 5 through 8[[Image:20060825CombinedSections5Through8AmplificationTrial1.jpg|thumb|right|pcr amplification of the purified product of the assembly of sections 5 through 8]].
**Lane 1: 2-log ladder
**Lane 2: 5-8AA
**Lane 3: 5-8AB
**Lane 4: 5-8AC
**Lane 5: 5-8 T7+ A
**Lane 6: 5-8 T7+ B
**Lane 7: 5-8 T7+ C
**Lane 8: 5-8 assembly product
**Lane 9: 5-9B
**Lane 10: 2-log ladder
**There seems to be a successful amplification in lane 7, but it does not seem to be the same size as the 5-8 assembly product.
====August 28, 2006====
*I ran a large [[Media:20060828AssembledSections567GelExtraction1.jpg|gel]] to isolate the pcr assembly product of the sections 5 - 7 fusion[[Image:20060828AssembledSections567GelExtraction1.jpg|thumb|right|gel isolation of the sections 5 - 7 assembly product]].
**Lane 1: 2-log ladder
**Lanes 2-3: 5-7G
**Lanes 4-5: 5-7H
**Lanes 6-8: 5-7I
**Lane 9: 2-log ladder
**I cut out the bands at the 3333bp position and put them in the fridge in 3 tubes labeled 5-7G, 5-7H, and 5-7I.
*I ran a PCR to amplify out sections 5, 6, 7, and 8 separately from the assembled sections 5-8 product.
**Master Mix:
***166.5uL nuclease-free water
***45uL 5x phusion hf buffer
***2.25uL 25mM dNTPs
***4.5uL 5-8ASS
**I put 48.5uL of MM into each of 4 tubes: 5-8 Amp 5, 5-8 Amp 6, 5-8 Amp 7, and 5-8 Amp 8.
**I put 0.5uL of section 5 forward primer and 0.5uL of section 5 reverse primer into 5-8 Amp 5.
**I put 0.5uL of section 6 forward primer and 0.5uL of section 6 reverse primer into 5-8 Amp 6.
**I put 0.5uL of section 7 forward primer and 0.5uL of section 7 reverse primer into 5-8 Amp 7.
**I put 0.5uL of section 8 forward primer and 0.5uL of section 8 reverse primer into 5-8 Amp 8.
**I added 0.5uL of phusion DNA polymerase to each of the 4 tubes.
**I ran the following pcr amplification program:
**#98.0*C for 30 seconds
**#98.0*C for 10 seconds
**#65.0*C for 30 seconds
**#72.0*C for 30 seconds
**#go to 2, 35 times
**#72.0*C for 10 minutes
**#4*C for 1 minute
**I placed the pcr tubes in the 4*C fridge when they finished. I will run a gel on these tomorrow.
====August 29, 2006====
*I ran a small [[Media:20060829Sections5678AssembledSectionCheckTrial1.jpg|gel]] on yesterday's PCR products[[Image:20060829Sections5678AssembledSectionCheckTrial1.jpg|thumb|right|pcr amplification of the individual sections within the sections 5 through 8 assembly]].
**Lane 1: 2-log ladder
**Lane 2: 5-8 Amp 5
**Lane 3: Sections 5 template
**Lane 4: 5-8 Amp 6
**Lane 5: Sections 6 template
**Lane 6: 5-8 Amp 7
**Lane 7: Section 7 template (from 7A)
**Lane 8: 5-8 Amp 8
**Lane 9: Section 8 template
**Lane 10: 2-log ladder
**The amplification products appear to be the same size as their original templates. This may mean that the assembly really did work. Tomorrow I will amplify the individual sections out of the sections 5 through 9 assembly.
**I ran out of section 6 and section 8 templates when loading the wells. I will need to make more.
*I gel extracted yesterday's gel isolates using a Qiagen extraction kit.
**5-7G: 310uL gel volume
***30uL final volume at 40.2ng/uL
**5-7H: 290uL gel volume
***30uL final volume at 12.7ng/uL
**5-7I: 380uL gel volume
***30uL final volume at 14.7ng/uL
====August 30, 2006====
*I ran a PCR to retry the amplification of sections 5 through 8 out of the 4 part assembly.
**Master Mix:
***240.5uL nuclease-free water
***65uL 5x phusion hf buffer
***3.25uL 25mM dNTPs
***3.25uL section 5 forward primer
***3.25uL section 8 reverse primer
**I put 48.5uL into each of 6 tubes: 5-8AD, 5-8AE, 5-8AF, 5-8 T7+ D, 5-8 T7+ E, and 5-8 T7+ F.
**I put 1uL of 5-8ASS into each of 3 tubes: 5-8AD, 5-8AE, and 5-8AF.
**I put 1uL of T7+ DNA into each of 3 tubes: 5-8 T7+ D, 5-8 T7+ E, and 5-8 T7+ F.
**I added 0.5uL phusion DNA polymerase to each of the 6 tubes.
**I ran the following amplification program:
**#98.0*C for 30 seconds
**#98.0*C for 10 seconds
**#65.0*C (column 1, 5-8AD and 5-8 T7+ D), 67.2*C (column 6, 5-8AE and 5-8 T7+ E), and 70.0*C (column 12, 5-8AF and 5-8 T7+ F) for 30 seconds
**#72.0*C for 4 minutes
**#go to 2, 35 times
**#72.0*C for 10 minutes
**#4*C for 1 minute
====September 1, 2006====
*I ran a small [[Media:20060901CombinedSections5Through9AmplificationTrial2.jpg|gel]] on Wednesdays' PCR amplification[[Image:20060901CombinedSections5Through9AmplificationTrial2.jpg|thumb|right|pcr amplification of sections 5 through 8 out of the combined sections 5 through 8 purified assembly product]].
**Lane 1: 2-log ladder
**Lane 2: 5-8 AD
**Lane 3: 5-8 AE
**Lane 4: 5-8 AF
**Lane 5: 5-8 T7+ D
**Lane 6: 5-8 T7+ E
**Lane 7: 5-8 T7+ F
**Lane 8: 5-8 ASS
**Lane 9: 5-9 B
**Lane 10: 2-log ladder
**I finally have a PCR product at the correct position. This indicates that my PCR assembly has most likely been working. I will need to try the same amplification on the sections 5 through 9 purified assembly product.
====September 6, 2006====
*I attempted to amplify sections 5 through 9 out of the purified sections 5 through 9 assembly.
**Master Mix:
***265.625uL nuclease-free water
***31.25uL 10x AccuPrime Pfx Reaction Mixture
***3.125uL section 5 forward primer
***3.125uL section 9 reverse primer
**I put 48.5uL of MM into each of 6 tubes: 5-9AD, 5-9AE, 5-9AF, 5-9 T7+ D, 5-9 T7+ E, and 5-9 T7+ F.
**I put 1uL of 5-9B into each of 5-9AD, 5-9AE, and 5-9AF.
**I put 1uL of T7+ DNA into each of 5-9 T7+ D, 5-9 T7+ E, and 5-9 T7+ F.
**I put 0.5uL of AccuPrimer Pfx DNA Polymerase into each of the 6 tubes.
**I ran the following amplification program:
**#95.0*C for 2 minutes
**#95.0*C for 15 seconds
**#55.0*C (column 1, 5-9AD and 5-9 T7+ D), 61.0*C (column 7, 5-9AE and 5-9 T7+ E), and 65.0*C (column 12, 5-9AF and 5-9 T7+ F) for 30 seconds
**#68.0*C for 5 minutes 15 seconds
**#go to 2, 35 times
**#4*C for 1 minute
====September 7, 2006====
*I attempted to amplify T7.2 sections 2, 4, 5, 6, and 7 together.
**Master Mix:
***379.25uL nuclease free water
***102.5uL 5x phusion hf buffer
***5.125uL 25mM dNTPs
**I added 47.5uL of MM to each of 10 tubes: 2A, 2B, 4A, 4B, 5A, 5B, 6A, 6B, 7A, and 7B.
**I added 0.5uL of construct 8 forward primer and 0.5uL of construct 8 reverse primer to each of tubes 2A and 2B.
**I added 0.5uL of construct 7 forward primer and 0.5uL of construct 7 reverse primer to each of tubes 4A and 4B.
**I added 0.5uL of construct 6 forward primer and 0.5uL of construct 6 reverse primer to each of tubes 5A and 5B.
**I added 0.5uL of construct 10 forward primer and 0.5uL of construct 10 reverse primer to each of tubes 6A and 6B.
**I added 0.5uL of construct 9 forward primer and 0.5uL of construct 9 reverse primer to each of tubes 7A and 7B.
**I added 1uL of template from well E2 (construct 8) to each of tubes 2A and 2B.
**I added 1uL of template from well D2 (construct 7) to each of tubes 4A and 4B.
**I added 1uL of template from well C2 (construct 6) to each of tubes 5A and 5B.
**I added 1uL of template from well G2 (construct 10) to each of tubes 6A and 6B.
**I added 1uL of template from well F2 (construct 9) to each of tubes 7A and 7B.
**I added 0.5uL of phusion DNA polymerase to each tube.
**I ran the following amplification program:
**#98.0*C for 30 seconds
**#98.0*C for 10 seconds
**#60.0*C (column 1 - 2A, 4A, 5A, 6A, and 7A) and 65.0*C (column 12 - 2B, 4B, 5B, 6B, and 7B) for 30 seconds
**#72.0*C for 30 seconds
**#go to 2, 35 times
**#72.0*C for 10 minutes
**#4*C for 1 minute
*I moved the constructs from Codon into new tubes - I labeled tubes containing T7.2 sections in black and tubes containing T7 sections in red.
**I moved T7.2 section 2 (construct 8) from well E2 into a 0.5mL PCR tube labeled "2."
**I moved T7.2 section 3 (construct 2) from well B1 into a 0.5mL PCR tube labeled "3."
**I moved T7.2 section 4 (construct 7) from well D2 into a 0.5mL PCR tube labeled "4."
**I moved T7.2 section 5 (construct 6) from well C2 into a 0.5mL PCR tube labeled "5."
**I moved T7.2 section 6 (construct 10) from well G2 into a 0.5mL PCR tube labeled "6."
**I moved T7.2 section 7 (construct 9) from well F2 into a 0.5mL PCR tube labeled "7."
**I moved T7.2 section 8 (construct 1) from well A1 into a 0.5mL PCR tube labeled "8."
**I moved construct 3 from well C1 into a 0.5mL PCR tube labeled "3."
**I moved construct 11 from well H2 into a 0.5mL PCR tube labeled "11."
**I moved construct 12 from well A2 into a 0.5mL PCR tube labeled "12."
====September 8, 2006====
*I ran a small [[Media:20060908T7-2Sections2456AmplificationTrial1.jpg|gel]] to check on my amplification of the codon constructs of T7.2 sections[[Image:20060908T7-2Sections2456AmplificationTrial1.jpg|thumb|right|pcr amplification of codon constructs 8, 7, 6, and 10]].
**Lane 1: 2-log ladder
**Lane 2: 2A
**Lane 3: 2B
**Lane 4: 4A
**Lane 5: 4B
**Lane 6: 5A
**Lane 7: 5B
**Lane 8: 6A
**Lane 9: 6B
**Lane 10: 2-log ladder
**The major products in lanes 2-3 and 6-9 seem to be the right size. Lanes 4-5 have the wrong pcr product. Every lane has multiple products in the wrong positions, which suggests a lot of unspecific binding. I may need to repeat the PCR with an increased annealing temperature.I also may need to PCR amplify section 6 separately so that I can shorten the extension time specially for that section.
====September 14, 2006====
*I ran a small [[Media:20060914CombinedSections5Through9AmplificationTrial3.jpg|gel]] on the amplification of sections 5 through 9 using accuprime pfx dna polymerase[[Image:20060914CombinedSections5Through9AmplificationTrial3.jpg|thumb|right|pcr amplification of sections 5 through 9 from the pcr assembled product and from T7+ DNA, using AccuPrime Pfx DNA Polymerase]].
**Lane 1: 2-log ladder
**Lane 2: 5-9 AD
**Lane 3: 5-9 T7+ D
**Lane 4: 5-9 AE
**Lane 5: 5-9 T7+ E
**Lane 6: 5-9 AF
**Lane 7: 5-9 T7+ F
**Lane 8: 5-8 ASS
**Lane 9: 5-9 C
**Lane 10: 2-log ladder
**The amplification using accuprime didn't work. I will need to optimize the use of accuprime pfx for our uses. In the meantime, I will attempt to amplify out sections 5 through 9 again using phusion dna polymerase.
====September 15, 2006====
*I ran a PCR to attempt to amplify sections 5 through 9 from the assembled product again.
**Master Mix:
***240.5uL nuclease-free water
***65uL 5x phusion hf buffer
***3.25uL 25mM dNTPs
***3.25uL section 5 forward primer
***3.25uL section 9 reverse primer
**I put 48.5uL into each of 6 tubes: 5-9AG, 5-9AH, 5-9AI, 5-9 T7+ G, 5-9 T7+ H, and 5-9 T7+ I.
**I put 1uL of 5-9C into each of 3 tubes: 5-9AG, 5-9AH, and 5-9AI.
**I put 1uL of T7+ DNA into each of 3 tubes: 5-9 T7+ G, 5-9 T7+ H, and 5-9 T7+ I.
**I added 0.5uL phusion DNA polymerase to each of the 6 tubes.
**I ran the following amplification program:
**#98.0*C for 30 seconds
**#98.0*C for 10 seconds
**#67.0*C (column 1, 5-9AG and 5-9 T7+ G), 70.1*C (column 7, 5-9AH and 5-9 T7+ H), and 72.0*C (column 12, 5-9AI and 5-9 T7+ I) for 30 seconds
Followed the Making T-broth Agar Plates protocol to make 40 T-broth agar plates. Sri taught me this method.
June 15, 2006
June 16, 2006
June 19, 2006
I used the Streaking Plates protocol to streak one plate with BL21 cells and one plate with IJ1126 cells. Sri taught me this method.
June 20, 2006
I used the Making Overnight Suspensions protocol to make two overnight suspensions of BL21 cells. Sri taught me this method.
June 21, 2006
I used the Freezing Away a Cell Stock protocol to make 2 stocks of BL21 cells. These BL21 stocks are located in the bottom shelf of the -80 storage, 2nd row from the top, 2nd column from the left, and are labeled with blue stickers.
I used the Making Plaques protocol to make 8 plates, which I labeled "Blank A," "Blank B," "10uL A," "10uL B," "100uL A," "100uL B," "200uL A," and "200uL B." I obtained my phage stock (10mL) from Sri's T7+ stock #1. His stock was created 9/28/2006 at 2.4e11 PFU/mL. I labeled this stock T7+ #1. I made 4 approximately 1:100 dilutions of my stock for use in creating plaques. The dilutions are as follows:
After incubating the eight plates for 4 hours, Sri and I spotted no plaques. About one and a half hours later, we found a plaque on the "100uL B" plate and the "200uL B" plate. This suggests that we diluted the phage stock too much. The phage stock may have diluted over time, skewing our value for the stock concentration. Also, we noticed that the agar on the plates was not very smooth. This may be due to the soft agar not being warm enough before being poured. We will try keeping the soft agar at a higher temperature before we pour it, to allow it more time to spread out over the plate before it solidifies. Sri walked me through the methods today.
June 22, 2006
Sri showed me how to make a new T7 stock using the Studier Lysate Prep protocol.
I used the Making Plaques protocol to make 8 new plates: Blank A, Blank B, 10uL A, 10uL B, 100uL A, 100uL B, 200uL A, and 200uL B. I obtained phage from my T7+ #1 stock. Because Sri and I found very few plaques after 4 1:100 dilutions of my stock yesterday, we decided today to make only 3 1:100 dilutions of my stock and use the final dilution to create plaques. The dilutions are as follows:
For the 200uL B plate, after adding the final phage dilution to one of the centrifuge tubes with culture, I forgot to vortex the mixture before adding it to the 3mL of soft agar. I also spilled a bit of the phage/BL21/soft agar mixture while pouring it onto its plate. After pouring the 8 plates, I noticed a black spec in the soft agar of the 10uL B plate.
These are the plaque counts after 4 hours of incubation:
Blank A: 0
Blank B: 0
10uL A: 0
10uL B: 2
100uL A: 8
100uL B: 6
200uL A: 21
200uL B: 44
These numbers seem somewhat inconsistent, especially between the two 200uL plates. Also, there is not enough data to confidently determine the original phage stock concentration. However, if I were to use this data, I would pick the plaque counts from the 100uL plates to calculate an original phage concentration of my stock. From these plaque counts, I arrived at a calculated stock concentration of 7e6 PFU/mL.
The weight plates had some plastic debris around the sides
I placed the T-broth agar in the autoclave at 5:30PM.
While the T-broth agar was in the autoclave, I made two overnight cultures of BL21 cells, using the Making Overnight Suspensions protocol.
I removed the T-broth agar at 9:00PM and poured 40 plates. I noticed black specs from the agar in 2 plates and I got one of the plate covers dirty. The black specs may be from the agar, as some of the T-broth agar on the side of the flask was burnt during autoclaving. I also noticed white, stringy debris in many of the plates. It looked like the same plastic debris from the weigh plates.
June 23, 2006
I checked last night's t-broth agar plates and found no contamination
I used last night's two overnight cultures find the titer again for the original T7+ #1 stock and also for the new T7+ stock that I created yesterday (I labled this stock T7+ #2). I used the Making Plaques protocol for these titers:
To titer the T7+ #1 stock, I decided on making a serial dilution of one 1:100 and three 1:10:
Dilution 1: 1:100 50uL stock in 5mL T-broth
(50uL)(1mL/1000uL)(7e6) = (5mL)(Concentration 1)
Concentration 1 = 7e4 PFU/mL
Dilution 2: 1:10 500uL dilution 1 in 4.5mL T-broth
The resulting final dilution should be 10 times more concentrated than last night's final dilution of the T7+ #1 stock. This would allow for more plaques to form on the plates, allowing us to more precisely measure the plaque forming units of the stock. I decided to create plaques using 50uL, 100uL, and 150uL of the final dilution, since I expected the best data to surround these amounts of phage dilution. The plates were labeled Blank A, Blank B, Blank C, 50uL A, 50uL B, 50uL C, 100uL A, 100uL B, 100uL C, 150uL A, 150uL B, and 150uL C.
To titer the T7+ #2 stock, I decided to make 5 1:100 dilutions of the stock in order to produce a broad range of dilutions to test for plaque forming units:
Dilution 1: 1:100 50uL stock in 5mL T-broth
Dilution 2: 1:100 50uL dilution 1 in 5mL T-broth
Dilution 3: 1:100 50uL dilution 2 in 5mL T-broth
Dilution 4: 1:100 50uL dilution 3 in 5mL T-broth
Dilution 5: 1:100 50uL dilution 4 in 5mL T-broth
I decided to use 100uL from each of the last three dilutions just to see which dilution would be optimal for observing plaque forming units. I labeled my plates Blank #1, Blank #2, 100uL Dilution 3 #1, 100uL Dilution 3 #2, 100uL Dilution 4 #1, 100uL Dilution 4 #2, 100uL Dilution 5 #1, and 100uL Dilution 5 #2.
I could not use the Blank #1 plate because the top agar layer would not stick to the bottom agar. I did notice some water on the bottom agar before I poured the soft agar. This may be the cause of non-sticking top layer. Also, I may have disrupted this layer when I tilted to the plate to check to see if the top layer had solidified.
I incubated both plaque groups at 2:15.
I checked both plaque groups at 6:15 and found that there were plaques on all the plates except for the blanks and the dilution 4 and 5 groups for T7+ #2. For this reason, I allowed the T7+ #2 group to incubate a while longer in the hope that visible plaques would form on plates 100uL Dilution 4 #1-2 and 100uL Dilution 5 #1-2.
I removed the 12 plates for T7+ #1 at 6:30 and found the following plaque counts:
Blank A: 0
Blank B: 0
Blank C: 0
50uL A: 73
50uL B: 80
50uL C: 91
100uL A: 151
100uL B: 197
100uL C: 178
150uL A: 323
150uL B: 345
150uL C: 254
The three plates 100uL A, 100uL B, and 150uL A each had streaks of plaque in the middle of the plate that made it difficult to tell plaques apart from each other. This may have been caused by water on the bottom agar before I poured the top agar.
At 7:30, I removed the 8 plates for T7+ #2 and found the following:
Blank #1: I could not use this plate. The top agar did not solidify completely and formed a thin layer of agar that stayed separate from the bottom layer.
Blank #2: 0 plaques
100uL Dilution 3 #1: Too many plaques were formed on this plate. Also, due to the extended amount of time that these plates were incubated, the plaques had grown very large and overlapped with each other, making it difficult to distinguish one from another.
100uL Dilution 3 #2: Too many plaques were formed on this plate. Also, due to the extended amount of time that these plates were incubated, the plaques had grown very large and overlapped with each other, making it difficult to distinguish one from another.
100uL Dilution 4: #1: 0
100uL Dilution 4: #2: 0
100uL Dilution 5: #1: 0
100uL Dilution 5: #2: 0
I suspect that problems that I encountered today (separated top layer of agar, streaks of plaque, no plaque) were due to the presence of water on many of the plates. For this reason, I removed the T-broth agar plates that I made last night from their plastic bag and placed them on the lab bench to dry overnight. Hopefully, this will help fix my problem.
I made two more BL21 overnight cultures using the Making Overnight Suspensions protocol. I will use these cultures to make another attempt at finding the titer of the T7+ #2 stock.
From the data for the T7+ #1 titer group, we can find an approximate titer for the T7+ #1 stock. I use the plaque counts from the 50uL group because I found no streaks of plaque in those groups and the numbers seem fairly close to each other:
I used the Making Plaques protocol to make 12 plates: Blank A, Blank B, Blank C, 50uL A, 50uL B, 50uL C, 100uL A, 100uL B, 100uL C, 150uL A, 150uL B, and 150uL C. I made 4 1:100 serial dilutions and used the final dilution as the phage stock for the plates. The dilutions were as follows:
Dilution 1: 1:100 50uL stock in 5mL T-broth
Dilution 2: 1:100 50uL dilution 1 in 5mL T-broth
Dilution 3: 1:100 50uL dilution 2 in 5mL T-broth
Dilution 4: 1:100 50uL dilution 3 in 5mL T-broth
I touched the lip of the test tube for the 100uL B plate and I noticed bubbles in the soft agar of the 100uL C plate. I also dropped the lid of the 150uL B plate.
I incubated all the plates at 12:30PM.
I took out the plates at 4:30 and found the following plaque counts:
Blank A: 0 (still found water on the agar)
Blank B: 0
Blank C: 0
50uL A: 7
50uL B: 9
50uL C: 9
100uL A: 11
100uL B: 22
100uL C: 12
150uL A: 34
150uL B: 0
150uL C: 15
The bubbles in the agar of the 100uL C plate made it difficult to count plaques. The 100uL B plate seems to be inconsistent with the 100uL A and 100uL C plates which suggests that my touching the lip of the test tube holding the soft agar had an effect on either the amount of plaque or the amount of bacteria. Also, the presence of no plaque on the 150uL B plate suggests that dropping the lid of the plate affected either the bacterial growth or the infection.
The plaque counts for the 50uL plates seem consistent, so we can use those numbers to calculate an approximate titer for the T7+ #2 stock:
I placed all the plates in the 4*C fridge at 5:30PM and left 5 unused plates out to continue drying.
June 26, 2006
Of the 5 plates left to dry, I bagged 4 of them and placed them in the 4*C fridge on my shelf. I may have contaminated one with my finger. I left this plate on the bench.
I made two overnight cultures of IJ1126 cells using the Making Overnight Suspensions protocol. I used LB instead of T-broth in the protocol. I placed the plates in the 37*C incubation room at 6:40PM.
Sri and I obtained two bottles of LB from the media room. One was used to make the overnights and is sitting on the lab bench. I placed the other bottle in the 37*C incubation room at 6:40PM.
June 27, 2006
I obtained my two overnights of IJ1126 and the warm LB.
I obtained two flasks with the OD tubes on the sides.
I filled each with 20mL warm LB.
I used sterile technique.
I added 20uL of IJ1126 culture to one of the flasks to make a 1:1000 dilution of the culture.
I used sterile technique.
I placed this flask in the 30*C shaking water bath
I used the Making T-broth Agar Plates protocol to make 1L of T-broth agar, which I autoclaved at 1:00PM. I removed the 2L flask from the autoclave and placed it in the 65*C water bath at 3:15PM. I poured 20 plates a short while later.
While pouring water into the 2L flask to mix the T-broth agar, I spilled a little water onto the side of the flask.
While I was taking the 20 plates out of their original bags, I dropped two. I'm not sure if the inside of the plates got exposed. These plates are marked "dropped."
While I was pouring the plates, I got agar onto the lids of three plates. These plates are marked "agar on lid." I poured too much agar into one of these plates (I labeled this one "too much agar"), which was probably the reason the agar got onto the lid. For the other too, I probably handled the plates too carelessly and splashed agar onto the sides of the plate and lid. I ran out of agar for my last plate, so one plate has very little agar (I labeled this plate "not enough agar"). While flaming my plates to pop bubbles, I melted the side of the one of the plates. This plate is labeled "melted plate."
Sri walked me through the "Preparation of Competent Cells for Phage Transfection" protocol as written in the Supplementary Information section of the "Refactoring Bacteriophage T7" paper.
Sri placed a bottle of 50mM CaCl2 on ice. He then obtained two plastic centrifuge tubes and pipetted all of the IJ1126 culture out of the flask and into one of the plastic centrifuge tubes. He used a beaker to hold that tube on the weight plate and tared the beaker and centrifuge tube together. Sri then removed the tube containing culture and replaced it with the other tube. He left this tube open (and included the cap in the beaker) and poured water into the tube until the mass balance read zero. Sri then used the SA600 rotor to centrifuge the tubes (emphasized tightening of both knobs on the centrifuge cover) at 5,000 RPM. We waited until the centrifuge was up to speed to set the timer to 5min.
After 5 minutes, we removed the tubes. Sri poured the supernatant of the culture-containing tube into the sink and touched the lip of the tube to a paper towel to take away remaining liquid. He resuspended the pellet in 10mL of 50mM CaCl2, which he then put on ice. The amount of 50mM CaCl2 should correspond to half of the volume of the starting culture. We allowed this new suspension to incubate in the ice bucket in the 4*C fridge for 30 minutes.
After 30 minutes of incubation, Sri centrifuged the cells again (and also weighed out a new balance centrifuge tube) at 5,000 RPM for 5 minutes, this time labeling the IJ1126 cells with a blue sticker because the liquids in the tubes now had the same color. After 5 minutes, he poured the supernatant into the sink and touched the lip of the tube to a paper towel to soak up remaining CaCl2. He resuspended the cells in 2mL of CaCl2 (corresponding to one-tenth of the original culture volume). He put this suspension back on ice and put the ice bucket in the 4*C fridge at 6:50PM. This suspension will incubate overnight and tomorrow, the cells should be ready for transfection.
June 28, 2006
I checked yesterday's T-broth agar plates for contamination.
I found 6 plates with small spots of rough texture. This could be contamination. I set these plates aside.
Sri walked me through the "Transfection of the Ligation Products" protocol as written in the Supplementary Information section of the "Refactoring T7 Bacteriophage" paper.
Sri placed all of the pipette tip boxes in the -20*C fridge to cool before use. We refilled yesterday's ice bucket with ice to keep the IJ1126 cell suspension cold. Sri obtained the phage DNA stock, which has a concentration of 7.67e8 molecules/uL. We calculated that 5.22uL of this stock would give 4e9 DNA molecules and that 1.30uL of this stock would give 1e9 molecules of DNA. Sri prepared 10 centrifuge tubes that were to have different amounts of phage DNA: 1) 1e9 molecules 2) 1e9 molecules 3) 4e9 molecules 4) 4e9 molecules 5) blank 6) 1e9 molecules 7) 1e9 molecules 8) 4e9 molecules 9) 4e9 molecules 10) b We then put 200uL of the IJ1126 culture into each tube (we worked quickly because it is very important to keep the culture suspension cold in this step). We then added the assigned amount of DNA to each centrifuge tube (Sri added to tubes 1-5, and I added to tubes 6-10). When performing this step, it is important to remember the following: a) be careful not to touch the lip of the DNA stock because it is easily contaminated, b) be careful not to get ice on the inside of the centrifuge tube lid because the ice may fall into the tube, c) always add DNA to the centrifuge tubes in numerical order to avoid confusion, and d) spin down the tube quickly before adding the DNA so the culture is at the bottom of the tube and will not leave the tube when it is opened. After adding the DNA, we allowed the DNA/IJ1126 mixture to incubate on ice for 30 minutes.
During this 30 minutes, I obtained soft agar from the 55*C incubator to pour into 10 small test tubes which were kept on a heat block. I microwaved the bottle of the soft agar for 50 seconds at 50% power. This caused the soft agar to boil. I pipetted 3mL soft agar into each of the 10 small test tubes on the heat block, paying special attention to sterile technique.
After the 30 minute incubation period, we heat shocked the cells. Sri worked with tubes 1-5, and I worked with tubes 6-10. We each labeled one plate for each of our tubes with the number corresponding to the tube. We heat shocked each sample one by one. For each sample, we 1) spun down the tube briefly, 2) used the 1000uL micropipette to suck up the whole mixture out of the tube, 3) ejected the mixture into a hot soft agar test tube (paying special attention to sterile technique), 4) tapped the test tube on the lab bench lightly to mix for 5 seconds, and 6) poured the test tube onto its corresponding plate. When I heat shocked my samples, I forgot to flame the lip of the hot agar test tube after adding the DNA/IJ1126 mixture for samples 6, 7, and 8. This may cause contamination in plates 6, 7, and 8. We placed our plates in the 37*C incubation room at 3:40PM to incubate for 3-5 hours.
During the lab meeting, Sri moved the plates from the 37*C incubation room to the 4*C cold room. We noted no growth on plate #7, which may mean that I added DNA to sample #6 a second time when I was supposed to add to #7. This is probably the case because some confusion about which samples I added DNA to arose during that step. I will avoid this in the future by always adding in numerical order. Plates 8 and 9 had a low efficiency. We moved the plates to the 4*C fridge.
I bagged 26 of my T plates and placed them in the 4*C fridge. I discarded 12. One of these had too little agar, one had too much, and one had melted plastic. I discarded the other nine because I suspected contamination within those plates.
I incubated these overnights and also placed a bottle of LB in the 37*C incubation room at 7:25PM.
June 29, 2006
I repeated the procedure that Sri showed me on June 27, 2006 for the preparation of competent cells for phage transfection with the exceptions detailed below.
I added 40uL of overnight culture instead of 20uL to get a 1:500 dilution of cells. I placed this in the 30*C shaking water bath at 10:30AM.
I retrieved the T-broth agar from the autoclave at 3:30PM. I could not retrieve the T-broth agar earlier because the building was evacuated due to a fire alarm. I placed the T-broth agar in the 65*C water bath.
I retrieved the IJ1126 culture from the shaking water bath at 4:10PM.
I observed a klett of 51.
I put the culture on ice immediately. Instead of pipetting the culture into a centrifuge tube, I flamed the lip of the flask and poured the culture directly into a centrifuge tube. I labeled this culture with a pink sticker. After centrifuging the culture, I resuspended the cells in 10mL of 50mM CaCl2 from Sri's stock. I allowed this to incubate for approximately 30 minutes.
During the 30 minutes incubation, I attempted to pour 40 plates of T-broth agar.
I did not pour 5 plates because I ran out of agar.
1 plate had way too much agar and 1 plate had way too little agar.
I noticed debris in 5 plates, probably from the agar.
I ended with 28 normal T-broth agar plates.
At 5:15PM, I centrifuged the IJ1126 suspension and resuspended the cells in 2mL of 50mM CaCl2 from the stock labeled "LYC 6/29/04." We could not use Sri's original stock because of a lab spill. I placed the 2mL IJ1126 suspension in the ice bucket and in the 4*C fridge. This will incubate overnight.
June 30, 2006
I repeated the procedure that Sri showed me on June 28,2006 for T7 transfection with changes detailed below.
I used the 7.47e8 molecules/uL T7 DNA stock for this transfection.
I setup ten samples: 1-8) 1e9 molecules (1.34uL stock), 9-10) blank.
The DNA stock ran out after I added DNA to samples 1, 2, and 3. For this reason, I used only 6 samples: 1-3) 1e9 and 4-6) blank.
During the 30 minute DNA/IJ1126 incubation, I pipetted 3mL of soft agar into each of 6 test tubes on a heat block.
During the 30 minutes incubation, I also checked yesterday's T plates. Of the plates I observed yesterday to be normal, I found 5 with possible contamination (funny texture in some spots on the agar), and 1 with way too much agar.
I poured my 6 samples into plates at 1:10PM.
The soft agar was not smooth when it solidified. The reason could be that I did not take my T-plates out of the 4*C fridge early enough and they did not have enough time to equilibrate to room temperature. The is the most likely cause of the uneven surfaces of solidified soft agar because the plates were still cool when I poured the soft agar.
I incubated the plates at 1:15PM in the 37*C incubation room.
I retrieved the T-broth agar at 4:35PM and immediately placed it in the 65*C water bath.
I retrieved my transfection plates at 4:40PM and counted plaques:
75
53
5
0
0
0
I found out that I ran out of DNA after adding DNA to sample 3. I sucked up the last bit of DNA stock for sample #3, which may have been less than 1.34uL since there was no DNA stock leftover. This is most likely what happened and would account for the extremely low plaque count in sample 3.
There were lots of bubbles in the top agar, which made it difficult to count plaques.
I retrieved the T-broth agar from the 65*C water bath and poured 37 plates at 5:20PM.
1 plate had way too much agar.
I ran out of agar while pouring my last plate, so that one plate has too little agar.
I must be generally pouring too much agar into each plate because in making the last two sets of T-broth agar plates, I have run out of agar before I could completely pour 40 plates.
July 1, 2006
I bagged the T-plates I made on June 29, 2006.
I found one with possible contamination (strange spot of texture in the agar).
I dropped one plate.
I placed a total of 20 T-plates in the 4*C fridge at 11:45AM.
I turned off the 30*C shaking water bath.
July 2, 2006
I bagged 22 plates from June 30 and put them in the 4*C fridge at 3:20PM.
I left 13 plates from June 30 out on the lab bench because they had spots of funny texture.
I placed a bottle of LB in the 37*C incubation room at 4:30PM.
July 6, 2006
I repeated the procedure that Sri showed me on June 27, 2006 for the preparation of competent cells for phage transfection.
I made two samples- each with 40uL of overnight culture to make 1:500 dilutions. I also included one blank for the klett calibration. I incubated these in the 30*C shaking water bath at 12:00PM.
I removed my samples at 5:35PM. I observed a klett of 50 and 52 for my two cell samples.
When transferring the cultures from the flasks to the centrifuge tubes, I flamed the lip of the flasks and simply poured the cultures directly into their centrifuge tubes. There is a chance for contamination in this step.
I placed my samples in the 4*C fridge at 7:10PM for an overnight incubation.
I worked more quickly this time around. My sterile technique seems to be good enough, so I believe that I can improve my experimental technique by working faster.
I flamed the pipette for too long and the pipette became very hot. When I sucked up the LB, it boiled and some LB shot into the filter of Sri's pipette-aid. I had to change the filter in the pipette-aid before transferring 5mL of LB into my two test tubes.
I placed these two cultures and a bottle of LB in the 37*C incubation room at 7:25PM.
July 7, 2006
I repeated the procedure from yesterday for the preparation of competent cells for phage transfection (two 20mL samples- each with 40uL of overnight culture for 1:500 dilutions).
I placed the two flasks in the 30*C shaking water bath at 12:10PM.
I repeated the procedure from June 28, 2006 for phage transfection.
I setup 9 tubes and 8 plates at 1:05PM (1 tube for water for the blanks):
1-3) 1e9 DNA molecules in the 50 klett IJ1126 cells
4-6) 1e9 DNA molecules in the 52 klett IJ1126 cells
7) blank with the 50 klett IJ1126 cells
8) blank with the 52 klett IJ1126 cells
I used this stock: T7+ CsCl2 DNA PREP 7-1-04 (~8e7 molecules/uL)
I calculated that I would need 12.5 uL of the stock for every 1e9 molecules of DNA.
I began the 30 minutes DNA/IJ1126 incubation on ice at 3:40PM.
I began the 3-5 hour incubation in the 37*C incubation room at 4:25PM.
I removed the two IJ1126 cultures from the shaking water bath at 5:50PM and I turned off the water bath. I observed kletts of 51 and 52.5. When transferring the cultures to centrifuge tubes, I flamed the lips of the flasks and poured directly. This introduces a chance for contamination. During this step, I dropped the cap of the tube with the klett 51 culture.
I began the 30 minute CaCl2 incubation at 6:25PM.
The final 2mL suspensions went into the 4*C fridge (in an ice bucket) at 7:20PM.
I removed the 8 plates from the 37*C incubation room at 7:30PM and counted the following plaques:
55
132
50
100
38
29
0
0
I noticed that the plaques were small. Next time, I will wait a bit longer before removing the plates from the incubation room. The plaques will then be bigger and easier to count. These 8 plates are now on my shelf in the 4*C fridge.
July 8, 2006
I repeated the procedure from June 28, 2006 for transfection. I used 12.5uL from a 8e7 molecule/uL stock for each sample to give 1e9 molecules of phage DNA for each test sample. I prepared 9 1.7 mL centrifuge tubes:
1-3) heat shock for 10 seconds
4-6) heat shock for 20 seconds
7-9) blank (instead of adding DNA, I added 12.5uL of water)
I placed 9 plates in the 37*C room before starting my 30 minute DNA/IJ1126 incubation to make them warm before pouring agar
I used the IJ1126 cells from the klett 52.5 culture because I dropped the cap of the klett 51 culture yesterday.
When adding DNA to tube #4, a small drop of DNA stock fell out of the pipette tip before the DNA was added.
I incubated the 9 plates in the 37*C room at 4:00PM.
I retrieved the 9 plates at 8:15PM and counted plaques:
18
41
19
0
19
40
0
0
0
In plate 1, there was some funny texture in the top agar that I think resulted from my pouring the soft agar. In plate 3, there were 4 plaques that were unusually large. In plates 4, 7, and 8, the soft agar layer had slipped off and clumped towards one side of the plate. I probably did not wait long enough for the top agar to solidify before inverting the plates and placing them in the 37*C incubation room. All 9 of these plates are now in the 4*C fridge for later examination.
July 10, 2006
Made one overnight of BL21 cells and one overnight of IJ1126 cells with LB, using the Making Overnight Suspensions protocol.
I incubated both overnights at 3:15PM.
When I first got to the 37*C incubation room, I found the door slightly open.
I placed these in the -80*C storage at 10:20AM with my BL21 stock.
I retrieved my othe two overnights at 11:15AM.
I put 40mL of LB into two armed flasks.
One of the pipettes had some debris inside.
I placed 1mL of overnight BL21 culture into one of the flasks.
I placed this flask in the 37*C shaking water bath at 11:30AM.
Sri and I obtained two 1L flasks at 12:05PM.
We poured approximately 240 - 250mL of LB directly into the flasks.
I put 20mL of culture in each flask from the armed flask and placed these flasks in the 37*C shaking water bath to bring the LB up to temp.
After the flasks had incubated in the shaking water bath for a few hours, Sri read on OD of 0.25.
He then added 2 drops of my phage stock to each flask.
When the flasks cleared, we added 11.69 grams of NaCl to each flask.
We swirled the flasks to mix the salt in.
Sri then poured the cultures straight into large centrifuge tubes.
We centrifuged these using the GS3 rotor for 10 minutes at 9000RPM.
We used water to balance the two tubes.
Sri then poured the supernatant into new large centrifuge tubes.
Sri put 26 grams of PEG into each tube. We shook the tubes to mix the PEG in.
We placed these tubes on ice for 1 hour.
During this hour, we got two small glass test tubes and poured CsCl2 step gradients.
Sri first pipetted 1mL of p=1.43 CsCl2 into each tube.
He then used a syringe (20g 1/2) to inject 1mL of p=1.53 CsCl2 in the bottom of each tube.
Sri used a syringe again (20g 1/2) to inject 1mL of p=1.62 CsCl2 to the bottom of each tube.
He used nitrile gloves because the CsCl2 was very concentrated.
He injected very slowly and then gradually sped up.
After the 1 hour incubation, we centrifuged the tubes at 5000 RPM for 15 minutes.
Sri poured out the supernatant and resuspended the pellet of one tube in 7mL of T7 buffer. He then took this suspension and used it to resuspend the pellet of the other tube.
Sri poured this new suspension into a 40mL centrifuge tube. We balanced the weight of this tube with another 40mL centrifuge tube.
We centrifuged these tubes for 10 minutes at 5000 RPM.
Sri then poured the supernatant (phage) into a cup.
Sri used a syringe to slowly drip the supernatant down the side of one of the test tubes with the CsCl2 layers to put the phage on top of the step gradient without disturbing the layers.
He put 5mL of phage in each tube.
We then obtained the test tube holding apparatus that goes with the SW41T1-4 rotor.
Sri used MilliQ water and a micropipette to slowly fill up each tube nearly to the lip with water. He did this on a balance to make sure the tubes were the same weight.
Sri loaded the test tubes into holders 1 and 4, placed the lids on all the holders, and screwed the lids in with a coin.
We then obtained the SW41T1-4 rotor and brought all of our apparatus to the ultracentrifuge.
We hooked the tube holders up to the metal bars in the rotor, aligned the pegs of the rotor with the centrifuge, and observed the revolution number to be 1602820.
Sri set the ultracentrifuge to speed: 30,000 RPM, time: 3 hours, and temperature: 4*C.
He set the Accel and Decel to "MAX."
He hit the vacuum button:
The "750" sign blinked and then went to 200.
Sri hit the start button.
The spin began at around 8:15PM.
I used yesterday's overnights to streak one plate with BL21 and one plate with IJ1126 using the Streaking Plates protocol.
I incubated these plates at 11:15PM, which means they should be ready by 2:15PM tomorrow.
Sri and I removed the step gradient tubes at 11:30PM.
We did not observe any phage bands.
Next time, we may mark the positions of the layers before centrifuging to help visualize the layers better.
We believe that there may have been leftover PEG in the T7 buffer step, which would cause the phage to pellet out of solution during the following centrifuge step. Also, the CsCl2 solutions may be a bit old. We may make new stocks.
I placed these in the shaking water bath at 12:15PM at 37*C.
I obtained two 1L bottles of LB.
I put one 1L bottle and one 500mL bottle in the 37*C incubation rom at 12:35PM.
Sri and I obtained these bottles and Sri poured 500mL of LB into each of two 2L flasks.
I pipetted 40mL of today's culture into each flask.
We incubated the flasks at 1:45PM in the 37*C room shaker.
I retrieved yesterday's streaked plates at 2:10PM.
There were very few individual colonies on either plate, which suggests that the culture I used was too concentrated. Later I will streak plates again, but using the -80 stocks, which have already been diluted with 40% glycerol.
I observed ODs of my flasks to be 0.36 and 0.42 at 3:25PM.
I added 5 drops of my phage stock to each flask.
I incubated these again at 3:27PM.
At 4:15, I retrieved the flasks (they had just cleared up), poured 29.22 grams of NaCl (1M) into each flask, mixed, and poured each culture into large centrifuge tubes. There was a small amount of culture leftover in each flask. I these tubes for 10 minutes at 9000RPM in the GS3 rotor.
After the spin, Sri and I very carefully poured the supernatant of these tubes into a large, wide 2L flask.
I placed the flask in the 4*C cold room at 5:00PM.
I streaked one more plate of BL21 cells from the -80 stock and one more plate of IJ1126 cells from the -80 stock using the Streaking Plates protocol.
I incubated these plates at 7:10PM, which means they should be ready by 10:10AM tomorrow.
July 13, 2006
I obtained my T7+ lysate, poured in 100g of PEG 8000, and mixed it in the 4*C cold room for several minutes. I then transferred the lysate to a 2L flask, which I put on ice and incubated in the cold room for 1 hour at 12:00PM.
I retrieved the flask at 1:00PM and poured the lysate into two 500mL centrifuge tubes. There was a little lysate leftover in the flask.
I centrifuged these tubes at 5000 RPM for 15 minutes in the GS3 rotor.
There was a lot of foam at the top of the bottle, which stuck to the seal of the cap.
I balanced the two tubes with water.
When the centrifuge finished, I poured out the supernatant.
I took some extra time to touch the lip of the tube to a paper towel to try to remove as much of the supernatant as possible, in order to reduce the amount of PEG leftover.
I resuspended one of the pellets with 7mL of T7 buffer, vortexed this tube thoroughly, then used the new suspension to resuspend the pellet of the 2nd tube.
I pipetted the suspension into a 40mL centrifuge tube.
I noticed that the final volume was a total of 9.25 mL, which meant that some extra liquid had been picked up in the two lysates. I added 10mL of extra T7 buffer to the 40mL centrifuge tube to decrease the concentration of any possible leftover PEG.
I spun this tube (with a balance) at 5000 RPM for 10 minutes at 2:40PM, using the SA-600 rotor.
After the spin, I poured the supernatant into a cup, parafilmed the cup, and placed it on ice.
I noticed a large amount of pellet in the centrifuge tube.
I poured a few practice step gradients using p = 1.43, 1.53, and 1.62 CsCl2.
I used 3mL syringes with 20 g 1/2 needles to create the step gradients.
When handling needles, it is very important to avoid people and to avoid pointing the tip outwards in order to reduce the risk of poking someone.
When I had finally made two good step gradients, I used a 10mL syringe with a 20 g 1/2 needle to slowly place 5mL of lysate on top of each gradient.
I used a 10mL syringe and a 20 g 1/2 needle again to fill the rest of each tube up nearly to the lip with T7 buffer this time, instead of water. These gradients were complete at 3:50PM.
Sri and I obtained the tube holders and placed the tubes in holders 1 and 4. Sri also checked the tubes on the mass balance to make sure they were the same weight. This is a very important step because small differences in weight can damage the ultracentrifuge.
Sri and I used the ultracentrifuge to spin down the lysate/step gradients.
The rotor has two holes on the side of the botton piece that marks the location of two stubs that point out of the bottom of the rotor. These two stubs connect with two holes in the centrifuge. This helps with more smoothly aligning the rotor.
We set the ultracentrifuge for speed: 30,000RPM, time: 5 hours, and temperature: 4*C. The acceleration and decceleration were set to "MAX."
Sri hit the vacuum button and we started the spin at 4:30PM.
I retrieved my two streaked plates from yesterday at 4:50PM. The BL21 plate had many individual colonies. The IJ1126 plate had more individual colonies, but they were smaller. I parafilmed these plates and placed them in the 4*C fridge.
I retrieved the centrifuge tubes at 9:45PM.
I observed 3 bands- the lowest one was the phage band (thin fine layer, whitish in color).
Sri showed me how to extract the phage from the 1st test tube and I extracted the phage from the 2nd test tube.
I used a 5mL syringe with a 26 g 3/8 needle.
I inserted the needle through the plastic, just below the phage band.
I tilted the needle up into the phage band and extracted slowly.
I ejected the phage solution into a 1.7mL centrifuge tube and placed it in the 4*C fridge.
July 14, 2006
I checked all the sequences for the PCR segments that we will work with. They all match either T7+ or T7.2 and all of the overlaps are exactly 30 base pairs long. There was a problem with segment T7.2rF_3173-3327u. When I initially aligned the sequence against the other segments, VectorNTI placed the sequence at position 2000 on the T7.2 sequence because a large portion of T7.2rF_3173-3327u is identical to the sequence at position 2000. This may be a problem for designing primers for PCR.
July 17, 2006
I designed primers for some sections of T7+ that Sri and I will PCR amplify. The list of primers can be found here.
I examined the melting temperatures, annealing temperatures, hairpin loops, and palindromes for each sequence.
I redesigned the primers for these sections. The new primers can be found here (Sri designed the first section).
This time, I focused primarily on matching the melting temperatures of each forward primer with its reverse primer. I did also try to keep all lengths within 20 - 25 base pairs.
I messed two up (1 had too much agar + bubbles, 1 had too little agar).
Some plates had uneven texture and visible solid agar.
There was still too much agar in all of my plates, which is why I ran out before pouring 40 plates.
The water bath was at 22.1*C when I placed the T-broth agar in after autoclaving because it had been off. When I removed the T-broth agar, the water temperature had risen to around 40*C. I suspect that the water bath not being up to temp probably cooled the agar too much, causing it to start solidifying while I was pouring.
I finished the plates at 3:15PM.
Sri walked me through a digestion of CsCl2 purified T7 DNA.
He used Bcl1, which creates at 8311 bp fragment and a 39.1 kb fragment.
He added 20uL of his stock to 20uL of nuclease-free water.
He used the nanodrop to observe a concentration of 1425.0 ng/uL for the mixture.
He wiped the lense with a kim-wipe, blanked with 1 uL of water, wiped again, read 1uL of DNA, and wiped again.
Sri added 40uL of water to dilute the DNA further.
This time, the nanodrop gave a concentration of 507.3 ng/uL.
Sri checked the NEB catalogue. He noted that Bcl1 is methylation sensitive.
Sri added the following together: 78uL DNA, 5uL Bcl1, 7uL nuclease-free water, and 10uL 10X NEB3 buffer.
5uL Bcl1 gives 75 units.
He placed the digestion in the 50*C heat block at 5:30PM for overnight digestion.
Sri walked me through the dialysis for our CsCl2 purified T7 phage from July 13, 2006.
Sri obtained the dialysis tubing from the 4*C fridge.
He put 500mL of T7 buffer in a 500mL beaker, which he put in the 4*C fridge at 5:45PM.
Sri pulled out some tubing (approximately 2 inches), clipped the end of it, and cut it off from the rest of the tubing.
Sri pipetted all the CsCl2 purified phage into the bag (dialysis tube closed off on the bottom with a dialysis clip).
He folded the other end and clipped it with another dialysis clip.
He dropped this in the 500mL beaker with T7 buffer.
The bag floated because he forgot to use weighted dialysis clips.
He tried to avoide bubbles in the bag.
Sri added a stir bar to the beaker.
We moved the stir plate to the 4*C room and began stirring the beaker at 5:50PM in the 4*C room. This will continue overnight.
Next time, we will remember to use weighted dialysis clips.
We should make more T7 buffer.
July 19, 2006
Sri removed our dialysis beaker from the 4*C cold room.
He squeezed the liquid out of the top and removed the clip.
He then pipetted the phage suspension into a 1.7mL eppindorf tube.
We received phenol today.
Always wear nitrile gloves and shoes when handling phenol.
The company we order from puts the phenol in a tightly sealed can. The plastic seal has to be cut off before you pry the lid off.
We put the crystallized phenol in a water bath set for 50*C and later turned the temperature up to 60*C.
We poured a 0.5% agar gel to run yesterday's digestion.
Sri mixed 0.35g of megabase agar in 70mL of TAE buffer.
He washed the gel apparatus, using some MilliQ water also.
We wore nitrile gloves for this because there may have been traces of ethidium bromide in the apparatus.
We placed the gel holder in the gel apparatus and checked the seal to make sure it was a good seal.
Sri microwaved the agar with the lid open slightly to dissolve the megabase agar in the buffer.
After pouring the agar into the gel holder, Sri put the comb for making lanes in- thin combs pointing down.
We left this apparatus at room temperature for the agar to cool and solidify.
Sri pipetted 50mL of phenol (after it melted) into a bottle under the hood.
He added 0.05 g of hydroxyquinoline to the phenol bottle.
He then added 50mL of 0.5M Tris-Cl.
We shook the bottle hard for 5 minutes and waited for the two layers to separate.
We aspirated the top layer.
We added 50mL more of 0.5M Tris-Cl.
The bottle cap was leaking, so we transferred the mixture to a new bottle.
We shook the bottle for 5 minutes, waited for the layers to separate, and then aspirated the top layer.
Sri then added 50mL of 0.1M Tris-Cl.
This time we put it on the stir plate for the duration of the lab meeting.
We mixed 9uL of our digested DNA with 27uL of nuclease-free water and 1uL of 10,000X SYBR Gold.
We mixed 1uL of the DNA ladder with 0.1uL of SYBER Gold and 4uL of water.
We added 8uL of 6x loading dye to the digested DNA tube and 1uL of 6x loading dye to the DNA ladder.
These mixtures were in small PCR tubes.
Sri filled the gel apparatus to the gel level with TAE buffer.
We added 2.3uL of digested DNA to each of 19 wells and 6uL of DNA ladder to one well.
We did this in the 4*C cold room.
Sri ran the gel at 20V (1-1.5V per cm) at 7:55PM.
I aspirated the aqueous layer of the phenol/tris-cl and read a pH of 7.0.
I added 50mL of 0.1M tris-cl, mixed on the stir plate, and aspirated the top layer again.
I read a pH of 7.0.
I added 50mL of 0.1M Tris-Cl again, mixed on the stir plate for 10 minutes, and aspirated the top layer again, this time leaving a small amount of aqueous layer behind.
I read of pH of 7.5 at 9:45PM.
I wrapped the bottle in tin foil and placed it in the 4*C fridge (the phenol is light sensitive).
I used pH paper to read the pH every time, using a aspirator tip to obtain a small amount of mixture each time.
I made two BL21 overnights at 9:25PM.
We ended up with one full bottle of phenol/chloroform waste tonight.
(Today's entry needs more detail)
July 20, 2006
At 12:10PM, Sri and I checked the gel using the gel imager.
We took the gel holder out of the apparatus and placed it in the gel imager (we used water to clean).
We chose the program "gene snap."
We chose "white light" to help us center the gel under the camera.
We chose "transilluminate" to help us visualize the bands.
The bands did not travel far.
We removed the gel holder and wiped down the imager with water.
We placed the gel holder back in the apparatus and turned the voltage up to 60.
Sri made aliquots of 25mM dNTPs.
Sri took the primer pellets and made 50uM primer suspensions in TE.
We setup a PCR tube for each section that we ordered primers for:
37uL water
10uL 5x phusion HF buffer
0.5uL of 25 mM dNTPs
0.5uL forward primer
0.5uL backward primer
1uL of template DNA
0.5uL phusion DNA polymerase
We first made a master mix:
337uL water
91uL buffer
4.55 uL dNTPs
We put 47.5 uL of master mix into each of 9 PCR tubes.
We ran out on the 9th tube, so Sri made the reaction mixture for that sample separately.
We used the thin-walled PCR tubes and set them up inside cold blocks.
We then added the 0.5 uL of each primer to each tube and the 1uL of template for each tube.
The last thing we added was the DNA polymerase.
Used the following DNA for templates:
"T7, 1-8311, 11/6/03"
I used the nanodrop to measure 10.6 ng/uL.
We used this template for sections 7 and 8.
"T7, 1122-6016, GI"
39.7 ng/uL
We used this template for sections 3, 4, 5, and 6.
"T7, 1-1122, GI"
15.7 ng/uL
We used this template for sections 1 and 2.
I diluted each of the templates 1:50 (1uL DNA, 49uL TE).
We used some T7 wildtype DNA as a template also:
"T7 DNA, 1363 ng/uL, CsCl2, 1/22/04, SK"
I diluted this 1:100 (1uL DNA in 99uL TE) and observed a concentration of 7.7 ng/uL.
I diluted the dilution again 1:10 (10uL DNA dilution in 90uL of TE) and observed the concentration to be 2.2 ng/uL.
We used this template for section 9.
We ran the PCR on the 9 tubes.
We put sections 1, 3, 4, 7, 8, and 9 in block A.
We ran this cycle for block A:
Initial Denaturation: 98*C for 30 seconds
35 cycles of the following:
Denature: 98*C for 10 seconds.
Anneal: 55*C for 30 seconds.
Extend: 72*C for 15 seconds.
Final Extension: 72*C 10 minutes.
4*C for 1 minute.
We put sections 2, 5,and 6 in block B.
We ran the same cycle, but with a 30 seconds extension time instead of 15 seconds.
We checked the gel again and observed lots of streaking.
We probably loaded too much DNA.
We threw the gel out in the SYBR Gold waste. We will try the gel again Monday.
I made two more BL21 overnights at 5:45PM.
I checked the PCR machine at 7:00PM.
Block A took 1:36:12 to run and block B took 1:43:28 to run.
I placed the tubes in a rack and put them in the 4*C fridge on my shelf.
More detail needed in this entry.
July 21, 2006
Got 1% agarose from the 55*C incubator.
Sri poured between 35-40 mL into a graduated cylinder.
Sri put the gel holder into the pouring block and poured the agarose into the holder.
He pipetted 1uL EtBr into the gel.
He stirred a little.
When doing this, keep the EtBr in its box.
Sri pushed the bubbles in the gel to the side with the comb.
Sri obtained a square of parafilm and flipped the parafilm over to expose the clean side.
He made 9 1uL dots on the parafilm with 6x loading buffer (red).
He added 5uL of PCR product to each dot.
He added 4uL of MilliQ water to each dot.
He put the solidifed gel (in its holder) in the gel box and filled the buffer to the gel surface level with TAE buffer.
He loaded the wells in the 4*C cold room.
We used the RZA gel box at 120V (12V per cm).
We checked the gel with the gel imager at 10:45AM.
I noticed that lanes from block B (2,5, and 6) were fine.
These samples were from block B (30 second annealing time at 55*C)
Lanes 1, 3, 4, 7, 8, and 9 had problems.
These lanes were from block A (15 second annealing time at 55*C)
I ran the lanes again at 100V (1:00PM).
I turned it down to 40V.
I turned it up to 80V at 1:30PM.
I turned it up to 100V at 1:40PM.
I turned the gel apparatus off at 2:00PM.
The marker had only run halfway.
I made 25:24:1 phenol:chloroform:isoamyl alcohol.
I added 25mL of phenol, 24mL of chloroform, and 1mL of isoamyl alcohol to a 200mL bottle under the hood.
I used isopentyl alcohol (same thing as isoamyl alcohol).
The chloroform leaked from the pipette really easily, so I do not think I got all 24 mL of chloroform into the miture (some dripped out while transferring from the stock to the mixture).
It was difficult to see the phenol/tris-cl layers, so I think that I grabbed some of the aqueous layer from the phenol.
It looks like a 10 second vs 5 second extension time doesn't make a difference for section 3.
For section 4, as the annealing temperature increases, the amount of incorrect product increases too. Does this mean that the primers are more specific to a different sequence?
For section 9, as the annealing temperature increases, the amount of expected product increases. This confirms Sri's idea that by raising the annealing temperature, the reaction favors the more specific complements.
July 25, 2006
I got the phenol from the 65*C water bath.
I put 50mL in a 200mL bottle (under the hood).
I put a stir bar in, and the phenol solidified really quickly.
I put the 50mL of phenol in the 65*C water bath to melt again.
I put the stock phenol back in the -20*C fridge.
After the 50mL of phenol melted, I added 0.05 grams of 8-quinolinole.
I stirred this on the stir plate (I brought it into the hood).
I added 50mL of 0.5M Tris-Cl pH~8.05 and stirred.
I let it separate, aspirated the top layer, and added 50mL of 0.5M Tris-Cl pH~8.05
I setup the same PCR as yesterday.
I made a different master mix:
370uL of nuclease-free water
100uL of 5X phusion HF buffer
5uL of 25mM dNTPs
10uL of template DNA (T7+)
Just before I added the DNA polymerase, someone began a PCR using one of the blocks, so I let my reaction mixtures sit in the 4*C fridge for a bit.
While my reaction mixtures sat in the fridge, I let the layers of the phenol/tris-cl separate, aspirated the top layer, and observed a pH of 6-6.5. I added 50mL of 0.5M Tris-Cl pH~8.05
Sri had me begin the PCR anyways without the 3rd PCR block.
I ran the following gradients on sections 4 and 9:
9A: column 6: 59.3*C
9B: column 7: 61.0*C
9C: column 10: 64.3*C
3A: column 5: 57.8*C
3B: column 8: 62.4*C
3C: column 12: 65*C
I ran samples 4A and 4B through the program with a 10 second annealing time.
I ran sample 4C through the program with a 5 second annealing time (I started this PCR a little later when the 3rd PCR block became available - 1:30PM).
I let the layers of the phenol/tris-cl separate, aspirated the aqueous layer, and observed a pH of 6.5
I added 50mL of 0.5M Tris-Cl again.
I repeated the above two steps three more times with the following observations:
The first time around, I broke the tip off of the aspirator and had to use another aspirator tip to pull the broken glass out of the phenol. I lost some volume because of this. I observed a pH of 7.
The second time around, I observed a pH of 7.
The third time around, I observed a pH of 7-7.5
I separated the layers again, aspirated and again observed a pH of 7-7.5.
For section 3, as the annealing temperature increased, it gave more of the expected product (1080bp).
For section 4, the shorter annealing times for all samples eliminated the unexpected product from the previous PCRs and gave only the expected 130bp product.
For section 9, the higher annealing temperatures gave more of the expected product (780bp), but the bands with expected product were not as strong as in trial 4. Next time I will try to work more quickly when making the reaction mixture so that my results will be more clean.
In the last two gels I ran, the DNA ladder smeared, which suggests that I need more practice in loading wells.
I let the layers of the phenol/tris-cl separate and I noticed that the aqueous layer was not as clear as before.
I aspirated this layer and observed a pH of 7.5-8.0
I used two other stir bars to get the original stir bar out of the bottle.
I wrapped the bottle in tin foil and placed it in the 4*C fridge (6:00PM).
Initialize the water thing by flicking it on, then off.
The resistance must rise to 18 ohms.
I ran a gel:
3x section 3 (from trial 5 - 3c)
3x section 5 (from trial 3)
3x section 7 (from trial 3)
I used a 2-log ladder this time.
The mixtures were as follows:
For the 2-log ladder, 3uL of 6x orangeG buffer + 2uL of ladder + 10uL of milliQ water
For section 3, 3uL of 6x orangeG buffer + 12uL DNA
For section 5, 3uL of 6x orangeG buffer + 12uL DNA
For section 7, 3uL of 6x orangeG buffer + 12uL DNA
I ran this at 12:55PM at 80V.
When the gel finished running, I cut out the bands with a razor.
I put saran wrap over the UV light table.
I made sure to turn the UV light to the lowest setting (UV damages DNA).
I was careful not to touch the gel slices to other pieces of gel.
I placed my gel slices in 1.7mL centrifuge tubes.
During group meeting, Austen suggested that I talk to Tom Knight about PCR fusion ideas (adding the sections one by one).
I used the Qiagen gel extraction kit to isolate my DNA:
I measured the weights of the gels:
0.17g for section 3
0.28g for section 5
0.26g for section 7
These numbers translated to:
Approx. 170uL of gel for section 3
Approx. 280uL of gel for section 5
Approx. 260uL of gel for section 7
I added 3x the gel volume of buffer QG for each tube:
510uL for section 3
840uL for section 5
780uL for section 7
I put the tubes in the 50*C heat block for 10 minutes.
I vortexed the tubes every 2 minutes.
I put 800uL of 100% isopropanol in a 1.7mL centrifuge tube.
I added an amount equal to the gel volume to each tube:
Section 3: 170uL
Section 5: 280uL
Section 7: 260uL
I vortexed the tubes.
I put 800uL of each tube into a gel extraction column.
I spun these down for 1 minute at 13,000 rpm.
I discarded the flow through.
I pipetted the rest of the liquid into each column.
I spun this down for another 1 minute and discarded the flow-through.
I added 0.5mL of buffer QG to each column.
I spun this down for 1 minute and discarded the flow-through.
I added 0.75mL of buffer PE to each tube.
I spun this down for 1 minute and discarded the flow-through.
I spun the column down again for 2 more minutes and discarded the flow through again.
I placed the columns in new 1.7mL centrifuge tubes.
I added 50uL of buffer EB to each tube and let it sit for 1 minute.
I spun these tubes for 1 minute, orientating the tubes so that the caps pointed away from the direction of rotation.
I took the columns out of the tubes. The DNA was in the bottom of the tube.
I used the nanodrop to observe the DNA concentrations:
Section 3: 1.3 ng/uL
Section 5: 12.8 ng/uL
Section 7: 7.5 ng/uL
I placed the tubes in the 4*C fridge.
July 27, 2006
I repeated the two PCR programs from July 20, 2006.
Initial Denaturation: 98*C for 30 seconds
35 cycles of the following:
Denature: 98*C for 10 seconds
Anneal: 55*C for 30 seconds
Extend: 72*C for 15 - 30 seconds
Final extension: 72*C for 10 minutes
4*C for 1 minute
I ran sections 2, 5, and 6 in the group with a 30 second extension time.
I ran sections 1, 7, and 8 in the group with a 15 second extension time.
I setup 5 PCR tubes for each section.
I made the following master mix:
592uL of nuclease-free water
160uL of phusion HF buffer
8uL of 25mM dNTPs
16uL of template DNA (T7+)
When I was adding the DNA polymerase, I ran out of the phusion DNA polymerase after the 3rd tube of section 7. I used VENT DNA polymerase for the last two tubes of section 7 and for section 8.
When the PCR finished, I ran a large gel on section 1 at 5:30PM at 120V in the 4*C cold room:
3uL of 6x OrangeG loading dye, 2uL of log-2 ladder, 10uL of water
11X (3uL of 6x OrangeG loading dye, 12uL of DNA)
I accidentally added 12uL of water to the ladder. The ladder bands may be slightly weaker because of this.
I cut out the 367bp bands from the gel at 7:30. I put these into 4 1.7mL centrifuge tubes, which I placed in the 4*C fridge. I will gel extract these tomorrow.
First 3 lanes: section 2 from column 1 of yesterday's PCR
Next 3 lanes: section 2 from column 8 of yesterday's PCR
Last 3 lanes: section 2 from column 12 of yesterday's PCR
Upon imaging the gel, we can see that the middle set worked, so we now have template for section 2.
I'm not sure if I switched the PCR tubes on accident after removing them from the PCR machine yesterday. I will need to repeat this PCR to find out which annealing temperature works. But for now, I have the template I need to work on the PCR fusion. I will remember to always label.
I ran a large gel at 120V to isolate this template:
Lanes 1 - 3: 3uL of OrangeG Loading Dye + 12uL of MilliQ water
Lane 4: 3uL of OrangeG Loading Dye + 2uL of 2-log ladder + 10uL of MilliQ water
Lanes 5-7: 3uL of OrangeG Loading Dye + 12uL of PCR product
Lane 8: 3uL of OrangeG Loading Dye + 2uL of 2-log ladder + 10uL of MilliQ water
Lanes 9-12: 3uL of OrangeG Loading Dye + 12uL of MilliQ water
I gel extracted the bands for sections 3, 4, and 9 from yesterday:
I split up the 2nd set of bands into two groups: B and C
3A: 280uL gel volume
30uL final volume at 16.4 ng/uL
3B: 340uL gel volume
30uL final volume at 19.2 ng/uL
3C: 210uL gel volume
30uL final volume at 16.0 ng/uL
3B: 330uL gel volume
50uL final volume at 13.6 ng/uL
4B: 250uL gel volume
50uL final volume at 12.1 ng/uL
9B: 260uL gel volume
50uL final volume at 8.6 ng/uL
3C: 180uL gel volume
30uL final volume at 15.8 ng/uL
4C: 200uL gel volume
30uL final volume at 14.6 ng/uL
9C: 230uL gel volume
30uL final volume at 18.5 ng/uL
The A group may not be usable because I may have switched some tubes around during gel extraction.
I transferred 1B to 1A and 1D to 1C to consolidate the DNA.
I transferred 7B to 7A and 7D to 7C to consolidate the DNA.
I put all of the templates from PCR in the -20*C fridge.
I cut the bands containing section 2 template from my large gel and placed it in a 1.7mL eppindorf tube, which is now in the 4*C fridge. I will gel extract this tomorrow.
I will also run the following PCR program tomorrow to practice combining 2 overlapping templates (sections 5 and 6).
Reaction Mixture:
37uL nuclease-free water
10uL 5x phusion hf buffer
0.5uL 25mM dNTPs
0.5uL section 5F primer
0.5uL section 6R primer
0.5uL section 5 template
0.5uL section 6 template
0.5uL phusion DNA polymerase
PCR program:
98.0*C for 30 seconds
98.0*C for 10 seconds
54.0*C to 65.0*C temperature gradient for 30 seconds
72.0*C for 30 seconds
98.0*C for 10 seconds
54.0*C to 65.0*C temperature gradient for 30 seconds
72.0*C for 50 seconds
Go to step 5, 35 times
72.0*C for 10 minutes
4*C for 1 minute
August 2, 2006
I ran a PCR to combine sections 5 and 6:
Master Mix:
148uL nuclease-free water
40uL phusion hf buffer
2uL 25mM dNTPs
2uL section 5 template
2ul section 6 template
2uL section 5F primer
2ul section 6R primer
I placed 49.5uL of this master mix into each of three tubes.
I added 0.5uL of phusion DNA polymerase to each of these tubes.
I ran the following PCR program:
98.0*C for 30 seconds
98.0*C for 10 seconds
55.0*C for 30 seconds
72.0*C for 30 seconds
98.0*C for 10 seconds
65.0*C for 30 seconds
72.0*C for 50 seconds
Go to step 5, 35 times
72.0*C for 10 minutes
4*C for 1 minute
While the PCR ran, I gel extracted yesterday's section 2 bands:
350uL gel volume
50uL final volume at 4.8 ng/uL
When the PCR finished, I ran a small gel containing 3 lanes for each of the 3 tubes2 part assembly with sections 5 and 6.
I made more 1% agarose:
5g Agarose in 500mL of TAE, microwaved for 4 minutes at 100% power.
The image from the gel shows a band close to 3kb, which indicates that the two section fusion worked.
August 3, 2006
I ran a PCR to try to combine sections 5, 6, and 7.
I made a master mix:
148uL nuclease-free water
40uL 5x phusion hf buffer
2uL 25mM dNTPs
2uL section 5 forward primer
2uL section 7 reverse primer
2uL section 5 template
2uL section 6 template
2uL section 7 template (from 7A stock)
I added 50uL of the master mix to each of 3 tubes.
I added 0.5uL of phusion DNA polymerase to each tube.
I labeled these tubes 5-7A, 5-7B, and 5-7C.
I placed 5-7A into column 1 (55.0*C annealing temperature in step 3 below), 5-7B into column 7 (61.0*C annealing temperature in step 3 below), and 5-7C into column 12 (65.0*C annealing temperature in step 3 below).
I ran the following program:
98.0*C for 30 seconds
98.0*C for 10 seconds
55.0*C - 65.0*C for 30 seconds
72.0*C for 20 seconds
98.0*C for 10 seconds
60.0*C - 70.0*C for 30 seconds
72.0*C for 40 seconds
98.0*C for 10 seconds
65.0*C - 75.0*C for 30 seconds
72.0*C for 55 seconds
go to step 8, 35 times
72.0*C for 10 minutes
4*C for 1 minute
When the PCR finished, I ran a small gel at 40Vgel results of the first attempt at making a 3 part assembly (sections 5 through 7).
I ran 3 lanes of 5-7A, 3 lanes of 5-7B, and 3 lanes of 5-7C.
The PCR did not work.
I may have let the DNA sit out for too long while making the reaction mixture and while pouring my gel.
I may not have let the gel solidify enough before running my samples.
I ran a new PCR.
This time, I switched to phusion GC buffer because it may be better for long sequences of DNA.
I also switched the 7 template from the 7A stock to the 7C stock.
I used 0.66uL of each template instead of 0.5uL.
I also added 0.75uL of phusion DNA polymerase to each tube, instead of 0.5uL.
This lead to a final reaction volume of 51.25uL.
I labeled my tubes 5-7D, 5-7E, and 5-7F.
I placed 5-7D in column 1 (55.0*C annealing temperature in step 3 below), 5-7E in column 5 (57.8*C annealing temperature in step 3 below), and 5-7F in column 8 (62.4*C annealing temperature in step 3 below).
I ran a new program:
98.0*C for 30 seconds
98.0*C for 10 seconds
55.0*C - 65.0*C for 30 seconds
72.0*C for 30 seconds
98.0*C for 10 seconds
65.0*C - 75.0*C for 30 seconds
72.0*C for 50 seconds
98.0*C for 10 seconds
65.0*C - 75.0*C for 30 seconds
72.0*C for 1 minute
go to step 8, 35 times
72.0*C for 10 minutes
4*C for 1 minute
I increased the extension times because in the previous PCR program, the extension times were not consistent with yesterday's PCR, which worked.
During this PCR, I ran another gelsecond gel image of the 3 part fusion attempt:
Lanes 1-2: 5-7A
Lanes 3-4: 5-7B
Lanes 5-6: 5-7C
Lane 7: 5-6A
Lane 8: 7A
Lane 9: 7C
Template stocks 7A and 7C are fine. The results of this gel confirm that the PCR program/reaction mixture did not work for 5-7A, 5-7B, and 5-7C.
There are faint bands below the 100bp marker.
When the new PCR finished, I placed the tubes in the 4*C fridge.
I ran a small gel on the afternoon's PCR2nd attempt at a 3 part assembly (with sections 5 through 7):
Lanes 1-2: 5-7D
Lanes 3-4: 5-7E
Lanes 5-6: 5-7F
Lane 7: 5-6B
Lane 8: 5-6C
The gel image shows again that the PCR didn't work. I did notice bands at the 1.5-2.0kb position and bands at the <100bp position. I will run a PCR tomorrow that involves adding the template in steps and a PCR to combine sections 6 and 7.
August 4, 2006
I ran a repeat of the PCR program from August 2nd but with sections 6 and 7, instead of 5 and 6.
Master Mix:
148uL nuclease-free water
40uL 5x phusion hf buffer
2uL 25mM dNTPs
2uL section 6 template
2uL section 7 template
2uL section 6f primer
2uL section 7r primer
I addeed 49.5uL of MM into each of 3 tubes and added 0.5uL phusion DNA polymerase to each tube.
I ran the following program:
98.0*C for 30 seconds
98.0*C for 10 seconds
55.0*C for 30 seconds
72.0*C for 30 seconds
98.0*C for 10 seconds
65.0*C for 30 seconds
72.0*C for 40 seconds
go to 5, 35 times
72.0*C for 10 minutes
4*C for 1 minute
I ran a new PCR that would allow me to add the template in steps:
Master Mix:
148uL nuclease-free water
40uL phusion hf buffer
2uL 25mM dNTPs
2uL section 5 template
2uL section 6 template
I added 48.5uL of master mix into each of 3 tubes labeled 5-7G, 5-7H, 5-7I.
I added 0.5uL phusion DNA polymerase to each tube.
I ran the following short program with the PCR machine:
98.0*C for 30 seconds
98.0*C for 10 seconds
55.0*C for 30 seconds
72.0*C for 30 seconds
go to 2, 1 time
4*C for 1 minute
I added 1uL of section 7 template (from 7A) to 5-7G
New volume: 50uL
I added 1.5uL of section 7 template (from 7A) to 5-7H
New volume: 50.5uL
I added 1.5uL of section 7 template (from 7A) to 5-7I
New volume: 50.5uL
I meant to add 0.5uL of section 7 template to 5-7G, 1uL to 5-7H, and 1.5uL to 5-7I, but I accidentally added 1.5uL to each of 5-7H and 5-7I.
I ran the following short program on the 3 tubes:
98.0*C for 30 seconds
98.0*C for 10 seconds
60.0*C for 30 seconds
72.0*C for 50 seconds
go to 2, 1 time
4*C for 1 minute
I added 0.5uL of section 5 forward primer to each tube and 0.5uL of section 7 reverse primer to each tube.
New volumes:
5-7G: 51uL
5-7H: 51.5uL
5-7I: 51.5uL
I ran the following long PCR program:
98.0*C for 30 seconds
98.0*C for 10 seconds
65.0*C for 30 seconds
72.0*C for 1 minute 10 seconds
go to 2, 35 times
72.0*C for 10 minutes
4*C for 1 minute
When both PCRs finished, I placed the tubes in the 4*C fridge. I will run a gel on these tomorrow to get the results.
August 7,2006
I ran a small gel at 120Vsuccessful 3 part fusion (sections 5 through 7).
Lane 1: 2-log ladder
Lanes 2-3: 5-7G
Lanes 4-5: 5-7H
Lanes 6-7: 5-7I
Lanes 8-9: 6-7A
Lane 10: 2-log ladder
The PCR appears to have worked. The band in lanes 2-7 looks to be just above the 3kb position, which agrees with the expected length (3393bp) of sections 5, 6, and 7 fused together. In lanes 8 and 9, the band is between the 2kb and 3kb position, which means that the fusion of sections 6 and 7 worked (2133bp).
The band in lanes 2-3 is much stronger than the bands in lanes 4-7, which suggests that it was better to only add 1uL of section 7 template than to add 1.5uL of section 7 template.
I ran a PCR to attempt to fuse sections 5, 6, 7, and 8:
Master Mix:
148uL nuclease-free water
40uL phusion hf buffer
2uL 25mM dNTPs
2uL section 5 template
2uL section 6 template
I added 48.5uL of master mix into each of 3 tubes.
I labeled these tubes 5-8A, 5-8B, 5-8C.
I added 0.5uL phusion DNA polymerase to each tube.
I ran the following short program with the PCR machine:
98.0*C for 30 seconds
98.0*C for 10 seconds
55.0*C for 30 seconds
72.0*C for 30 seconds
go to 2, 1 time
4*C for 1 minute
I added 0.5uL of section 7 template (from 7A) to 5-8A
New volume: 49.5uL
I added 1uL of section 7 template (from 7A) to 5-8B
New volume: 50uL
I added 1.5uL of section 7 template (from 7A) to 5-8C
New volume: 50.5uL
I ran the following short program on the 3 tubes:
98.0*C for 30 seconds
98.0*C for 10 seconds
60.0*C for 30 seconds
72.0*C for 50 seconds
go to 2, 1 time
4*C for 1 minute
I added 0.5uL of section 8 template to 5-8A
New volume: 50uLuL
I added 1uL of section 8 template to 5-8B
New volume: 51uL
I added 1.5uL of section 8 template to 5-8C
New volume: 52uL
I ran the following short program on the 3 tubes:
98.0*C for 30 seconds
98.0*C for 10 seconds
60.0*C for 30 seconds
72.0*C for 1 minute 10 seconds
go to 2, 1 time
4*C for 1 minute
I added 0.5uL of section 5 forward primer to each tube and 0.5uL of section 8 reverse primer to each tube.
New volumes:
5-8A: 51uL
5-8B: 52uL
5-8C: 53uL
I ran the following long PCR program:
98.0*C for 30 seconds
98.0*C for 10 seconds
65.0*C for 30 seconds
72.0*C for 1 minute 30 seconds
go to 2, 35 times
72.0*C for 10 minutes
4*C for 1 minute
When this PCR finished running, I ran a small gel at 120Vsections 5 through 8 assembly product.
Lane 1: 2-log ladder
Lanes 2-3: 5-8A
Lanes 4-5: 5-8B
Lanes 6-7: 5-8C
Lanes 8-9: section 8 template
Lane 10: 2-log ladder
The expected length for a successful fusion of sections 5, 6, 7, and 8 was 4459bp. I observed strong bands around the 4kb and 5kb markers in lanes 6 and 7, and light bands at the same position in lanes 4 and 5. This suggests that adding more template between PCR steps improves the PCR fusion. Tomorrow, I will run the gel longer, or with a different marker, to clarify the positions of these bands.
August 8, 2006
I realize that yesterday, I may have accidentally been adding 1.5uL of each template to 5-8A and 0.5uL of each template to 5-8C. This may mean that it is better to add less template between PCR steps.
I ran a PCR to attempt to fuse together all 9 T7+ alpha sections.
I made a master mix:
148uL nuclease-free water
40uL phusion hf buffer
2uL 25mM dNTPs
2uL section 1 template (from 1A)
2uL section 2 template
I added 48.5uL of master mix to each of three tubes which I labeled 1-9A, 1-9B, and 1-9C.
I added 0.5uL phusion DNA polymerase to each tube.
New Volumes:
1-9A: 49uL
1-9B: 49uL
1-9C: 49uL
I ran the following short PCR program:
98.0*C for 30 seconds
98.0*C for 10 seconds
55.0*C for 30 seconds
72.0*C for 30 seconds
go to 2, 1 time
4*C for 1 minute
I added 1.5uL of section 3 template (from 3A) to 1-9A, 1uL to 1-9B, and 0.5uL to 1-9C.
New Volumes:
1-9A: 50.5uL
1-9B: 50uL
1-9C: 49.5uL
I ran the following short PCR program:
98.0*C for 30 seconds
98.0*C for 10 seconds
65.0*C for 30 seconds
72.0*C for 50 seconds
go to 2, 1 time
4*C for 1 minute
I added 1.5uL of section 4 template (from 4A) to 1-9A, 1uL to 1-9B, and 0.5uL to 1-9C.
New Volumes:
1-9A: 52uL
1-9B: 51uL
1-9C: 50uL
I ran the following short PCR program:
98.0*C for 30 seconds
98.0*C for 10 seconds
65.0*C for 30
72.0*C for 1 minute
go to 2, 1 time
4*C for 1 minute
I added 1.5uL of section 5 template to 1-9A, 1uL to 1-9B, and 0.5uL to 1-9C.
New Volumes:
1-9A: 53.5uL
1-9B: 52uL
1-9C: 50.5uL
I ran the following short PCR program:
98.0*C for 30 seconds
98.0*C for 10 seconds
65.0*C for 30 seconds
72.0*C for 1 minute 20 seconds
go to 2, 1 time
4*C for 1 minute**I added 1.5uL of section 6 template to 1-9A, 1uL to 1-9B, and 0.5uL to 1-9C.
New Volumes:
1-9A: 55uL
1-9B: 53uL
1-9C: 51uL
I ran the following short PCR program:
98.0*C for 30 seconds
98.0*C for 10 seconds
65.0*C for 30 seconds
72.0*C for 1 minute 40 seconds
go to 2, 1 time
4*C for 1 minute
I added 1.5uL of section 7 template (from 7A) to 1-9A, 1uL to 1-9B, and 0.5uL to 1-9C.
New Volumes:
1-9A: 56.5uL
1-9B: 54uL
1-9C: 51.5uL
I ran the following short PCR program:
98.0*C for 30 seconds
98.0*C for 10 seconds
65.0*C for 30 seconds
72.0*C for 2 minutes
go to 2, 1 time
4*C for 1 minute
I added 1.5uL of section 8 template to 1-9A, 1uL to 1-9B, and 0.5uL to 1-9C.
New Volumes:
1-9A: 58uL
1-9B: 55uL
1-9C: 52uL
I ran the following short PCR program:
98.0*C for 30 seconds
98.0*C for 10 seconds
65.0*C for 30 seconds
72.0*C for 2 minutes 20 seconds
go to 2, 1 time
4*C for 1 minute
I added 1.5uL of section 9 template (from 9A) to 1-9A, 1uL to 1-9B, and 0.5uL to 1-9C.
New Volumes:
1-9A: 59.5uL
1-9B: 56uL
1-9C: 52.5uL
I ran the following short PCR program:
98.0*C for 30 seconds
98.0*C for 10 seconds
65.0*C for 30 seconds
72.0*C for 2 minutes 40 seconds
go to 2, 1 time
4*C for 1 minute
I added 1uL of section 1 forward primer and 0.5uL of section 9 reverse primer to each tube.
There is no band at the 8520bp position (expected product of the fusion of sections 1 through 9) in any lane. There is a light band between the 5kb and 6kb position in lanes 2 and 3. Tomorrow I will run a gel on all of my gel extracts to confirm that my template stocks are okay.
There is smearing in the 2-log ladder, but not in the 1kb ladder. This suggests that there may be something wrong with the 2-log ladder stock.
August 9, 2006
I ran a PCR to attempt to fuse sections 5, 6, 7, 8, and 9:
Master Mix:
148uL nuclease-free water
40uL phusion hf buffer
2uL 25mM dNTPs
2uL section 5 template
2uL section 6 template
I added 48.5uL of master mix into each of 3 tubes.
I labeled these tubes 5-9A, 5-9B, 5-9C.
I added 0.5uL phusion DNA polymerase to each tube.
I ran the following short program with the PCR machine:
98.0*C for 30 seconds
98.0*C for 10 seconds
55.0*C for 30 seconds
72.0*C for 30 seconds
go to 2, 1 time
4*C for 1 minute
I added 0.5uL of section 7 template (from 7A) to 5-9A
New volume: 49.5uL
I added 1uL of section 7 template (from 7A) to 5-9B
New volume: 50uL
I added 1.5uL of section 7 template (from 7A) to 5-9C
New volume: 50.5uL
I ran the following short program on the 3 tubes:
98.0*C for 30 seconds
98.0*C for 10 seconds
60.0*C for 30 seconds
72.0*C for 50 seconds
go to 2, 1 time
4*C for 1 minute
I added 0.5uL of section 8 template to 5-9A
New volume: 50uL
I added 1uL of section 8 template to 5-9B
New volume: 51uL
I added 1.5uL of section 8 template to 5-9C
New volume: 52uL
I ran the following short program on the 3 tubes:
98.0*C for 30 seconds
98.0*C for 10 seconds
60.0*C for 30 seconds
72.0*C for 1 minute 10 seconds
go to 2, 1 time
4*C for 1 minute
I added 0.5uL of section 9 template (from 9A) to 5-9A
New volume: 50.5uL
I added 1uL of section 9 template (from 9A) to 5-9B
New volume: 52uL
I added 1.5uL of section 9 template (from 9A) to 5-9C
New volume: 53.5uL
I ran the following short program on the 3 tubes:
98.0*C for 30 seconds
98.0*C for 10 seconds
60.0*C for 30 seconds
72.0*C for 1 minute 30 seconds
go to 2, 1 time
4*C for 1 minute
I added 0.5uL of section 5 forward primer to each tube and 0.5uL of section 9 reverse primer to each tube.
gel showing the 5 part assembly (sections 5 through 9)
Lane 1: 2-log ladder
Lanes 2-3: 5-9A
Lanes 4-5: 5-9B
Lanes 6-7: 5-9C
Lanes 8-9: 9A
Lane 10: 1kb ladder
Samples 5-9B and 5-9C had stronger bands at the 5239bp position (expected length of sections 5 through 9 combined). This suggests that it is better to add more template between PCR steps.
Experimental factors to consider:
Ratio of templates: to increase our chances for the intermediate PCR steps to create the combined template, we should try adding increasing amounts of DNA template between PCR steps.
Ratio of the reaction mixture: since the reaction volume changes throughout the process, we may need to experiment with increased amounts of buffer, dNTPs, DNA polymerase, and primers.
Rounds of PCR: we will need to see what the optimal number of rounds is for each intermediate short PCR to more efficiently or quickly get the combined sections template.
Annealing temperature and extension time per additional template: I have been adding a standard 20 seconds to the extension time each time a template is added. This may be too much for certain sections.
Buffer: we should try using GC buffer, which is supposedly better for long DNA sequences.
I realize that in adding up the combined lengths of my sequences, I forgot to take into account the 30bp overlaps between each of the 9 T7+ alpha sections. My calculated lengths should actually be a bit shorter - I will recalculate these values later.
August 16, 2006
I ran a PCR to amplify the sections 5-9 fusion pcr from last week.
Master Mix:
148uL nuclease-free water
40uL 5x phusion hf buffer
2uL 25mM dNTPs
4uL sections 5-9 pcr fusion product (from 5-9C)
2uL section 5 forward primer
2uL section 9 reverse primer
I added 49.5uL of MM to each of three tubes labeled 5-9 PCR 1, 5-9 PCR 2, 5-9 PCR 3.
I added 0.5uL of phusion DNA polymerase to each tube.
gel image of the attempted amplification of sections 5 through 9 from the sections 5 through 9 assembly product
Lane 1: 2-log ladder
Lanes 2-3: 5-9 PCR 1
Lanes 4-5: 5-9 PCR 2
Lanes 6-7: 5-9 PCR 3
Lanes 8-9: 5-9C
Lane 10: 1kb ladder
The amplifications of 5-9C did not work. There are also many other bands in the PCR. We will figure out what those bands are later. Tomorrow I will run a gel on 5-9C and 5-9B to gel isolate the sections 5-9 combined template.
I ran a PCR to amplify the sequence from section 2 through section 9 on the T7+ DNA. This will test the range of phusion DNA polymerase.
Master Mix:
120.25uL nuclease-free water
32.5uL 5x phusion hf buffer
1.625uL 25mM dNTPs
3.25uL T7+ DNA
1.625uL section 2 forward primer
1.625uL section 9 reverse primer
I added 49.5uL of MM to each of three tubes labeled 2-9A, 2-9B, and 2-9C.
I added 0.5uL of phusion DNA polymerase to each tube.
I ran the following program on a gradient block:
98.0*C for 30 seconds
98.0*C for 10 seconds
55.0*C (column 1, 2-9A), 59.3*C (column 6, 2-9B), and 65.0*C (column 12, 2-9C) for 30 seconds
72.0*C for 2 minutes 20 seconds
go to 2, 35 times
72.0*C for 10 minutes
4*C for 1 minute
I ran a gel to test the quality of the template for each section.
gel image of each section's template
Lane 1: 2-log ladder
Lane 2: Section 1 Template (from 1A)
Lane 3: Section 2 Template
Lane 4: Section 3 Template (from 3A)
Lane 5: Section 4 Template (from 4A)
Lane 6: Section 5 Template
Lane 7: Section 6 Template
Lane 8: Section 7 Template (from 7A)
Lane 9: Section 8 Template
Lane 10: Section 9 Template (from 9A)
The lanes were clean and had bands in all the correct positions.
We will need to make more of sections 6 and 8 template.
gel isolation of the sections 5 through 9 assembly product
Lane 1: 2-log ladder
Lanes 2-4: 5-9B
Lanes 5-6: 5-9C
While the gel ran, I ran a PCR assembly on sections 1, 2, and 3.
I made a master mix:
129.5uL nuclease-free water
35uL 5x phusion hf buffer
1.75uL 25mM dNTPs
1.75uL section 1 template (from 1A)
1.75uL section 2 template
I may have accidentally added 2uL each of the dNTPs and the templates, instead of 1.75uL.
I added 48.5uL of MM into each of three tubes: 1-3A, 1-3B, and 1-3C.
I added 0.5uL of phusion DNA polymerase to each tube for a final volume of 49uL in each tube.
I ran the following extension program on the three tubes:
98.0*C for 30 seconds
98.0*C for 10 seconds
55.0*C for 30 seconds
72.0*C for 30 seconds
go to 2, 1 time
4*C for 1 minute
I added 0.5uL of section 3 template (from 3A) to 1-3A, 1uL to 1-3B, and 1.5uL to 1-3C.
New volumes: 1-3A: 49.5uL, 1-3B: 50uL, and 1-3C: 50.5uL.
I ran another extension program on the tubes:
98.0*C for 30 seconds
98.0*C for 10 seconds
60.0*C for 30 seconds
72.0*C for 40 seconds
go to 2, 1 time
4*C for 1 minute
I added 1uL of section 1 forward primer and 0.5uL of section 3 reverse primer to each tube.
Final volumes: 1-3A: 51uL, 1-3B: 51.5uL, and 1-3C: 52uL.
I ran a final PCR step to amplify the combined sections 1 - 3.
98.0*C for 30 seconds
98.0*C for 10 seconds
65.0*C for 30 seconds
72.0*C for 1 minute
go to 2, 35 times
72.0*C for 10 minutes
4*C for 1 minute
When my gel of the section 5-9 fusion finished running, I used the Qiagen Gel Extraction kit to purify the combined sections 5-9 band.
5-9B: 350uL gel volume
30uL final volume at 6.4ng/uL
5-9C: 250uL gel volume
30uL final volume at 5.7ng/uL
I ran a gel on yesterday's PCR product of the sections 2 through 9 amplification.
gel results of the amplification of sections 2 through 9 off of T7 wildtype DNA
Lane 1: 2-log ladder
Lanes 2-4: 2-9A
Lanes 5-7: 2-9B
Lanes 8-10: 2-9C
I observed no bands at the 7943bp position (expected length). The PCR of long sequences needs further optimization.
I ran a gel on the PCR assembly of sections 1, 2, and 3 from earlier today.
gel results of the PCR assembly of sections 1 through 3
The gel showed no bands at the 3091bp position (expected length of assembly). The extension times for each assembly step may not be long enough. I may need to design a new procedure to accomodate the small sizes of sections 1 and 3 (367bp and 130bp).
August 18, 2006
I ran a PCR assembly to attempt to put together sections 1 and 2.
Master Mix:
129.5uL nuclease-free water
35uL 5x phusion hf buffer
1.75uL 25mM dNTPs
1.75uL section 1 template
1.75uL section 2 template
I added 48.5uL of MM to each of three tubes: 1-2A, 1-2B, and 1-2C.
I added 0.5uL phusion DNA polymerase
I ran the following short assembly program:
98.0*C for 30 seconds
98.0*C for 10 seconds
55.0*C for 30 seconds
72.0*C for 30 seconds
go to 2, 1 time
4*C for 1 minute
I added 1uL of section 1 forward primer and 0.5uL of section 2 reverse primer to each of the three tubes.
Final volumes: 50.5uL
I ran the following PCR program:
98.0*C for 30 seconds
98.0*C for 10 seconds
65.0*C for 30 seconds
72.0*C for 40 seconds
go to 2, 35 times
72.0*C for 10 minutes
4*C for 1 minute
Sri will take out my PCR at 3:00PM.
August 21, 2006
I ran a small gel on the results of the sections 1 and 2 assembly from Friday. gel of results of the sections 1 and 2 assembly
Lane 1: 2-log ladder
Lanes 2-3: 1-2A
Lanes 4-5: 1-2B
Lanes 6-7: 1-2C
Lane 8: Section 1 Template (from 1A)
Lane 9: Section 2 Template
Lane 10: 2-log ladder
The gel showed bands around the 750bp position. Our expected length for the section 1 plus section 2 assembly was 2041bp.
I PCR amplified my sections 5 through 9 assembly from August 9, 2006. I will use my gel extract from August 17, 2006.
I made a Master Mix:
259uL nuclease-free water
70uL 5x phusion hf buffer
3.5uL 25mM dNTPs
3.5uL section 9 reverse primer
I put 48uL of MM into each of 6 tubes.
I put 1uL of T7+ DNA into each of 3 tubes labeled 2-9D, 2-9E, and 2-9F.
I put 0.5uL of section 2 forward primer into each of these tubes.
In the other 3 tubes, I put 1uL of assembled sections 5-9 and labeled these tubes 5-9 AA, 5-9 AB, 5-9 AC.
I put 0.5uL of section 5 reverse primer into each of these 3 tubes.
I put 0.5uL of phusion DNA polymerase into each of the 6 tubes.
I ran the following PCR program:
98.0*C for 30 seconds
98.0*C for 10 seconds
65.0*C for 30 seconds
72.0*C for 2 minutes (2-9D, 5-9AA), 3 minutes (2-9E, 5-9AB), and 4 minutes (2-9F, 5-9AC)
go to 2, 35 times
72.0*C for 10 minutes
4*C for 1 minute
These PCRs will run overnight.
I should test a section 4 + 5 assembly as a comparison to the section 1 + 2 assembly. I think there may be a problem with trying to fuse two DNA segments that are very different in size.
The sections 1 + 2 + 3 assembly and the sections 1 through 9 assembly may not have worked because of:
the size differences between sections 1 and 2, and 4 and 5
unspecific binding by the section 3 reverse primer
unspecific binding by the section 1 forward primer
I should also test the an assembly with sections 2 + 3 and sections 4 + 5 because on my very first attempt at PCR amplification of the sections from the T7+ DNA, all these sections had a second PCR product which was not the correct size.
I may want to try using more assembly rounds for the sections 1 + 2 and 4 + 5 assemblies.
August 22, 2006
I ran a PCR assembly on sections 4 and 5.
Master Mix:
129.5uL nuclease-free water
35uL 5x phusion hf buffer
1.75uL 25mM dNTPs
1.75uL section 4 template (from 4A)
1.75uL section 5 template
I added 48.5uL of MM to each of three tubes: 4-5A, 4-5B, and 4-5C.
I added 0.5uL phusion DNA polymerase
I ran the following short assembly:
98.0*C for 30 seconds
98.0*C for 10 seconds
50.0*C for 30 seconds
72.0*C for 30 seconds
go to 2, 1 time
4*C for 1 minute
I added 0.5uL of section 4 forward primer and 0.5uL of section 5 reverse primer to each of the three tubes.
Final volumes: 50uL
I ran the following PCR program:
98.0*C for 30 seconds
98.0*C for 10 seconds
65.0*C for 30 seconds
72.0*C for 30 seconds
go to 2, 35 times
72.0*C for 10 minutes
4*C for 1 minute
During the PCR, I ran a small gel on yesterday's PCR amplification of sections 2 through 9 from the T7+ DNA.
gel results of yesterday's PCR amplficication of sections 2 through 9 from the T7 wildtype DNA
Lane 1: 2-log ladder
Lanes 2-4: 2-9A
Lanes 5-7: 2-9B
Lanes 8-10: 2-9C
The gel did not show any bands above 3kb.
When the first PCR finished, I ran a new PCR assembly of sections 2 and 3.
Master Mix:
129.5uL nuclease-free water
35uL 5x phusion hf buffer
1.75uL 25mM dNTPs
1.75uL section 2 template
1.75uL section 3 template (from 3A)
I added 48.5uL of MM to each of three tubes: 2-3A, 2-3B, and 2-3C.
I added 0.5uL phusion DNA polymerase
I ran the following short assembly:
98.0*C for 30 seconds
98.0*C for 10 seconds
50.0*C for 30 seconds
72.0*C for 40 seconds
go to 2, 1 time
4*C for 1 minute
I added 0.5uL of section 2 forward primer and 0.5uL of section 3 reverse primer to each of the three tubes.
Final volumes: 50uL
I ran the following PCR program:
98.0*C for 30 seconds
98.0*C for 10 seconds
65.0*C for 30 seconds
72.0*C for 1 minute
go to 2, 35 times
72.0*C for 10 minutes
4*C for 1 minute
During this PCR, I ran a small gel on yesterday's PCR amplification of sections 2 through 9 from the T7+ DNAgel results of yesterday's PCR amplficication of the section 5 through 9 assembly gel extract.
Lane 1: 2-log ladder
Lanes 2-3: 5-9AA
Lanes 4-5: 5-9AB
Lanes 6-7: 5-9AC
Lanes 8-9: 5-9B
Lane 10: 2-log ladder
Lanes 2-7 showed no bands at the position of the band from lanes 8 and 9. Tomorrow I will try amplifying the section 5 through 8 assembly. I will also try amplifying sections 5-6, 5-7, 5-8, and 5-9 from wildtype DNA.
August 23, 2006
I ran a PCR assembly on sections 3 and 4.
Master Mix:
129.5uL nuclease-free water
35uL 5x phusion hf buffer
1.75uL 25mM dNTPs
1.75uL section 3 template (from 3A)
1.75uL section 4 template (from 4A)
I added 48.5uL of MM to each of three tubes: 3-4A, 3-4B, and 3-4C.
I added 0.5uL phusion DNA polymerase
I ran the following short assembly:
98.0*C for 30 seconds
98.0*C for 10 seconds
65.0*C for 30 seconds
72.0*C for 20 seconds
go to 2, 1 time
4*C for 1 minute
I added 0.5uL of section 3 forward primer and 0.5uL of section 4 reverse primer to each of the three tubes.
Final volumes: 50uL
I ran the following PCR program:
98.0*C for 30 seconds
98.0*C for 10 seconds
65.0*C for 30 seconds
72.0*C for 20 seconds
go to 2, 35 times
72.0*C for 10 minutes
4*C for 1 minute
I ran a large gel on my PCR assembly product of sections 5 through 8gel isolation of the assembly product of the sections 5 through 8 pcr fusion.
Lane 1: 2-log ladder
Lanes 2-3: 5-8C
I used the Qiagen Gel Extraction Kit to isolate the sections 5 though 8 assembly product.
5-8ASS 230uL gel volume
30uL final volume at 16.4ng/uL
August 24, 2006
I ran a PCR to amplify sections 5 through 8 off of the sections 5 through 8 4 part assembly.
Master Mix:
259u nuclease-free water
70uL 5x phusion hf buffer
3.5uL 25mM dNTPs
3.5uL section 5 forward primer
3.5uL section 8 reverse primer
I put 48.5uL into each of 6 tubes labeled 5-8AA, 5-8AB, 5-8AC, 5-8 T7+ A, 5-8 T7+ B, and 5-8 T7+ C.
I put 1uL of the gel extracted sections 5 through 8 assembly product into each of tubes 5-8AA, 5-8AB, and 5-8AC.
I put 1uL of T7+ DNA into each of tubes 5-8 T7+ A, 5-8 T7+ B, and 5-8 T7+ C.
I added 0.5uL of phusion DNA polymerase to each of the six tubes.
I ran the following PCR program:
98.0*C for 30 seconds
98.0*C for 10 seconds
65.0*C for 30 seconds
72.0*C for 2 minutes (5-8AA and 5-8 T7+ A), 3 minutes (5-8AB and 5-8 T7+ B), and 4 minutes (5-8AC and 5-8 T7+ C).
go to 2, 35 times
72.0*C for 10 minutes
4*C for 1 minute
I ran a small gel on the sections 4 and 5 assembly from August 22, 2006pcr assembly of sections 4 and 5.
Lane 1: 2-log ladder
Lanes 2-3: 4-5A
Lanes 4-5: 4-5B
Lanes 6-7: 4-5C
Lane 8: Section 4 template (from 4A)
Lane 9: Section 5 template
Lane 10: 2-log ladder
It appears that the assembly may have worked, but it is too hard to tell, since the expected product is not much bigger than section 5 itself. I will have to run this gel again, and for longer, in order to look for any separation between lanes 2 through 7 and lane 9.
I ran a small gel on the sections 2 and 3 assembly from August 22, 2006pcr assembly of sections 2 and 3.
Lane 1: 2-log ladder
Lanes 2-3: 2-3A
Lanes 4-5: 2-3B
Lanes 6-7: 2-3C
Lane 8: Section 2 template
Lane 9: Section 3 template (from 3A)
Lane 10: 2-log ladder
The expected product of the assembly is 2754bp. There is a strong band in each of lanes 2-7 that is near the 3kb position, which indicates that this assembly worked.
August 28. 2006
I ran a small gel on the pcr assembly of sections 3 and 4 from August 23, 2006gel results from a pcr assembly of sections 3 and 4.
Lane 1: 2-log ladder
Lanes 2-3: 3-4A
Lanes 4-5: 3-4B
Lanes 6-7: 3-4C
Lane 8: Section 3 template (from 3A)
Lane 9: Section 4 template (from 4A)
Lane 10: 2-log ladder
It is difficult to tell the assembly bands apart from the section 3 template band, with respect to size. I will have to run this gel again, but longer.
I ran a small gel on yesterday's pcr amplification of the gel isolated pcr assembly product of sections 5 through 8pcr amplification of the purified product of the assembly of sections 5 through 8.
Lane 1: 2-log ladder
Lane 2: 5-8AA
Lane 3: 5-8AB
Lane 4: 5-8AC
Lane 5: 5-8 T7+ A
Lane 6: 5-8 T7+ B
Lane 7: 5-8 T7+ C
Lane 8: 5-8 assembly product
Lane 9: 5-9B
Lane 10: 2-log ladder
There seems to be a successful amplification in lane 7, but it does not seem to be the same size as the 5-8 assembly product.
August 28, 2006
I ran a large gel to isolate the pcr assembly product of the sections 5 - 7 fusiongel isolation of the sections 5 - 7 assembly product.
Lane 1: 2-log ladder
Lanes 2-3: 5-7G
Lanes 4-5: 5-7H
Lanes 6-8: 5-7I
Lane 9: 2-log ladder
I cut out the bands at the 3333bp position and put them in the fridge in 3 tubes labeled 5-7G, 5-7H, and 5-7I.
I ran a PCR to amplify out sections 5, 6, 7, and 8 separately from the assembled sections 5-8 product.
Master Mix:
166.5uL nuclease-free water
45uL 5x phusion hf buffer
2.25uL 25mM dNTPs
4.5uL 5-8ASS
I put 48.5uL of MM into each of 4 tubes: 5-8 Amp 5, 5-8 Amp 6, 5-8 Amp 7, and 5-8 Amp 8.
I put 0.5uL of section 5 forward primer and 0.5uL of section 5 reverse primer into 5-8 Amp 5.
I put 0.5uL of section 6 forward primer and 0.5uL of section 6 reverse primer into 5-8 Amp 6.
I put 0.5uL of section 7 forward primer and 0.5uL of section 7 reverse primer into 5-8 Amp 7.
I put 0.5uL of section 8 forward primer and 0.5uL of section 8 reverse primer into 5-8 Amp 8.
I added 0.5uL of phusion DNA polymerase to each of the 4 tubes.
I ran the following pcr amplification program:
98.0*C for 30 seconds
98.0*C for 10 seconds
65.0*C for 30 seconds
72.0*C for 30 seconds
go to 2, 35 times
72.0*C for 10 minutes
4*C for 1 minute
I placed the pcr tubes in the 4*C fridge when they finished. I will run a gel on these tomorrow.
August 29, 2006
I ran a small gel on yesterday's PCR productspcr amplification of the individual sections within the sections 5 through 8 assembly.
Lane 1: 2-log ladder
Lane 2: 5-8 Amp 5
Lane 3: Sections 5 template
Lane 4: 5-8 Amp 6
Lane 5: Sections 6 template
Lane 6: 5-8 Amp 7
Lane 7: Section 7 template (from 7A)
Lane 8: 5-8 Amp 8
Lane 9: Section 8 template
Lane 10: 2-log ladder
The amplification products appear to be the same size as their original templates. This may mean that the assembly really did work. Tomorrow I will amplify the individual sections out of the sections 5 through 9 assembly.
I ran out of section 6 and section 8 templates when loading the wells. I will need to make more.
I gel extracted yesterday's gel isolates using a Qiagen extraction kit.
5-7G: 310uL gel volume
30uL final volume at 40.2ng/uL
5-7H: 290uL gel volume
30uL final volume at 12.7ng/uL
5-7I: 380uL gel volume
30uL final volume at 14.7ng/uL
August 30, 2006
I ran a PCR to retry the amplification of sections 5 through 8 out of the 4 part assembly.
Master Mix:
240.5uL nuclease-free water
65uL 5x phusion hf buffer
3.25uL 25mM dNTPs
3.25uL section 5 forward primer
3.25uL section 8 reverse primer
I put 48.5uL into each of 6 tubes: 5-8AD, 5-8AE, 5-8AF, 5-8 T7+ D, 5-8 T7+ E, and 5-8 T7+ F.
I put 1uL of 5-8ASS into each of 3 tubes: 5-8AD, 5-8AE, and 5-8AF.
I put 1uL of T7+ DNA into each of 3 tubes: 5-8 T7+ D, 5-8 T7+ E, and 5-8 T7+ F.
I added 0.5uL phusion DNA polymerase to each of the 6 tubes.
I ran the following amplification program:
98.0*C for 30 seconds
98.0*C for 10 seconds
65.0*C (column 1, 5-8AD and 5-8 T7+ D), 67.2*C (column 6, 5-8AE and 5-8 T7+ E), and 70.0*C (column 12, 5-8AF and 5-8 T7+ F) for 30 seconds
72.0*C for 4 minutes
go to 2, 35 times
72.0*C for 10 minutes
4*C for 1 minute
September 1, 2006
I ran a small gel on Wednesdays' PCR amplificationpcr amplification of sections 5 through 8 out of the combined sections 5 through 8 purified assembly product.
Lane 1: 2-log ladder
Lane 2: 5-8 AD
Lane 3: 5-8 AE
Lane 4: 5-8 AF
Lane 5: 5-8 T7+ D
Lane 6: 5-8 T7+ E
Lane 7: 5-8 T7+ F
Lane 8: 5-8 ASS
Lane 9: 5-9 B
Lane 10: 2-log ladder
I finally have a PCR product at the correct position. This indicates that my PCR assembly has most likely been working. I will need to try the same amplification on the sections 5 through 9 purified assembly product.
September 6, 2006
I attempted to amplify sections 5 through 9 out of the purified sections 5 through 9 assembly.
Master Mix:
265.625uL nuclease-free water
31.25uL 10x AccuPrime Pfx Reaction Mixture
3.125uL section 5 forward primer
3.125uL section 9 reverse primer
I put 48.5uL of MM into each of 6 tubes: 5-9AD, 5-9AE, 5-9AF, 5-9 T7+ D, 5-9 T7+ E, and 5-9 T7+ F.
I put 1uL of 5-9B into each of 5-9AD, 5-9AE, and 5-9AF.
I put 1uL of T7+ DNA into each of 5-9 T7+ D, 5-9 T7+ E, and 5-9 T7+ F.
I put 0.5uL of AccuPrimer Pfx DNA Polymerase into each of the 6 tubes.
I ran the following amplification program:
95.0*C for 2 minutes
95.0*C for 15 seconds
55.0*C (column 1, 5-9AD and 5-9 T7+ D), 61.0*C (column 7, 5-9AE and 5-9 T7+ E), and 65.0*C (column 12, 5-9AF and 5-9 T7+ F) for 30 seconds
68.0*C for 5 minutes 15 seconds
go to 2, 35 times
4*C for 1 minute
September 7, 2006
I attempted to amplify T7.2 sections 2, 4, 5, 6, and 7 together.
Master Mix:
379.25uL nuclease free water
102.5uL 5x phusion hf buffer
5.125uL 25mM dNTPs
I added 47.5uL of MM to each of 10 tubes: 2A, 2B, 4A, 4B, 5A, 5B, 6A, 6B, 7A, and 7B.
I added 0.5uL of construct 8 forward primer and 0.5uL of construct 8 reverse primer to each of tubes 2A and 2B.
I added 0.5uL of construct 7 forward primer and 0.5uL of construct 7 reverse primer to each of tubes 4A and 4B.
I added 0.5uL of construct 6 forward primer and 0.5uL of construct 6 reverse primer to each of tubes 5A and 5B.
I added 0.5uL of construct 10 forward primer and 0.5uL of construct 10 reverse primer to each of tubes 6A and 6B.
I added 0.5uL of construct 9 forward primer and 0.5uL of construct 9 reverse primer to each of tubes 7A and 7B.
I added 1uL of template from well E2 (construct 8) to each of tubes 2A and 2B.
I added 1uL of template from well D2 (construct 7) to each of tubes 4A and 4B.
I added 1uL of template from well C2 (construct 6) to each of tubes 5A and 5B.
I added 1uL of template from well G2 (construct 10) to each of tubes 6A and 6B.
I added 1uL of template from well F2 (construct 9) to each of tubes 7A and 7B.
I added 0.5uL of phusion DNA polymerase to each tube.
I ran the following amplification program:
98.0*C for 30 seconds
98.0*C for 10 seconds
60.0*C (column 1 - 2A, 4A, 5A, 6A, and 7A) and 65.0*C (column 12 - 2B, 4B, 5B, 6B, and 7B) for 30 seconds
72.0*C for 30 seconds
go to 2, 35 times
72.0*C for 10 minutes
4*C for 1 minute
I moved the constructs from Codon into new tubes - I labeled tubes containing T7.2 sections in black and tubes containing T7 sections in red.
I moved T7.2 section 2 (construct 8) from well E2 into a 0.5mL PCR tube labeled "2."
I moved T7.2 section 3 (construct 2) from well B1 into a 0.5mL PCR tube labeled "3."
I moved T7.2 section 4 (construct 7) from well D2 into a 0.5mL PCR tube labeled "4."
I moved T7.2 section 5 (construct 6) from well C2 into a 0.5mL PCR tube labeled "5."
I moved T7.2 section 6 (construct 10) from well G2 into a 0.5mL PCR tube labeled "6."
I moved T7.2 section 7 (construct 9) from well F2 into a 0.5mL PCR tube labeled "7."
I moved T7.2 section 8 (construct 1) from well A1 into a 0.5mL PCR tube labeled "8."
I moved construct 3 from well C1 into a 0.5mL PCR tube labeled "3."
I moved construct 11 from well H2 into a 0.5mL PCR tube labeled "11."
I moved construct 12 from well A2 into a 0.5mL PCR tube labeled "12."
September 8, 2006
I ran a small gel to check on my amplification of the codon constructs of T7.2 sectionspcr amplification of codon constructs 8, 7, 6, and 10.
Lane 1: 2-log ladder
Lane 2: 2A
Lane 3: 2B
Lane 4: 4A
Lane 5: 4B
Lane 6: 5A
Lane 7: 5B
Lane 8: 6A
Lane 9: 6B
Lane 10: 2-log ladder
The major products in lanes 2-3 and 6-9 seem to be the right size. Lanes 4-5 have the wrong pcr product. Every lane has multiple products in the wrong positions, which suggests a lot of unspecific binding. I may need to repeat the PCR with an increased annealing temperature.I also may need to PCR amplify section 6 separately so that I can shorten the extension time specially for that section.
September 14, 2006
I ran a small gel on the amplification of sections 5 through 9 using accuprime pfx dna polymerasepcr amplification of sections 5 through 9 from the pcr assembled product and from T7+ DNA, using AccuPrime Pfx DNA Polymerase.
Lane 1: 2-log ladder
Lane 2: 5-9 AD
Lane 3: 5-9 T7+ D
Lane 4: 5-9 AE
Lane 5: 5-9 T7+ E
Lane 6: 5-9 AF
Lane 7: 5-9 T7+ F
Lane 8: 5-8 ASS
Lane 9: 5-9 C
Lane 10: 2-log ladder
The amplification using accuprime didn't work. I will need to optimize the use of accuprime pfx for our uses. In the meantime, I will attempt to amplify out sections 5 through 9 again using phusion dna polymerase.
September 15, 2006
I ran a PCR to attempt to amplify sections 5 through 9 from the assembled product again.
Master Mix:
240.5uL nuclease-free water
65uL 5x phusion hf buffer
3.25uL 25mM dNTPs
3.25uL section 5 forward primer
3.25uL section 9 reverse primer
I put 48.5uL into each of 6 tubes: 5-9AG, 5-9AH, 5-9AI, 5-9 T7+ G, 5-9 T7+ H, and 5-9 T7+ I.
I put 1uL of 5-9C into each of 3 tubes: 5-9AG, 5-9AH, and 5-9AI.
I put 1uL of T7+ DNA into each of 3 tubes: 5-9 T7+ G, 5-9 T7+ H, and 5-9 T7+ I.
I added 0.5uL phusion DNA polymerase to each of the 6 tubes.
I ran the following amplification program:
98.0*C for 30 seconds
98.0*C for 10 seconds
67.0*C (column 1, 5-9AG and 5-9 T7+ G), 70.1*C (column 7, 5-9AH and 5-9 T7+ H), and 72.0*C (column 12, 5-9AI and 5-9 T7+ I) for 30 seconds












































{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
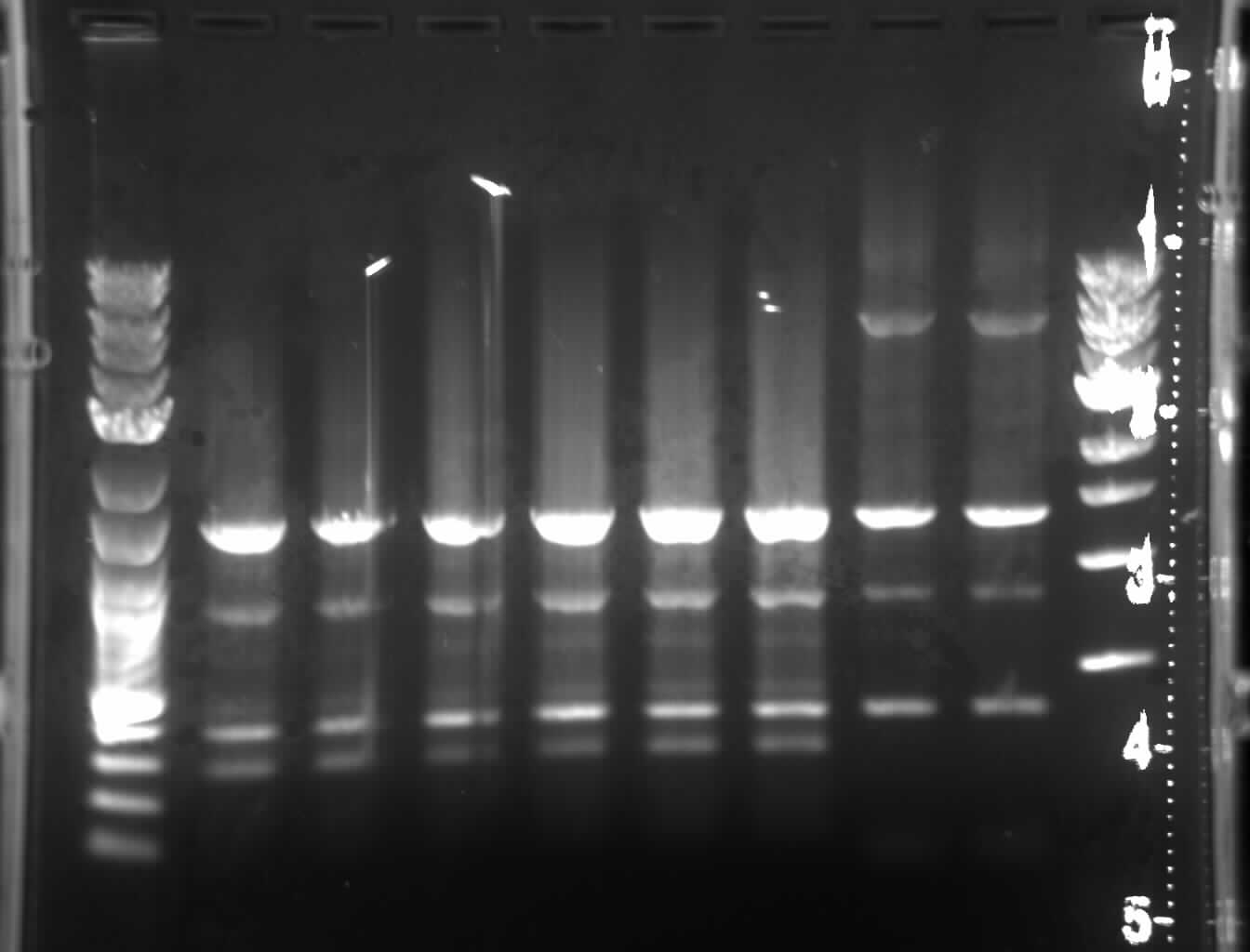{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
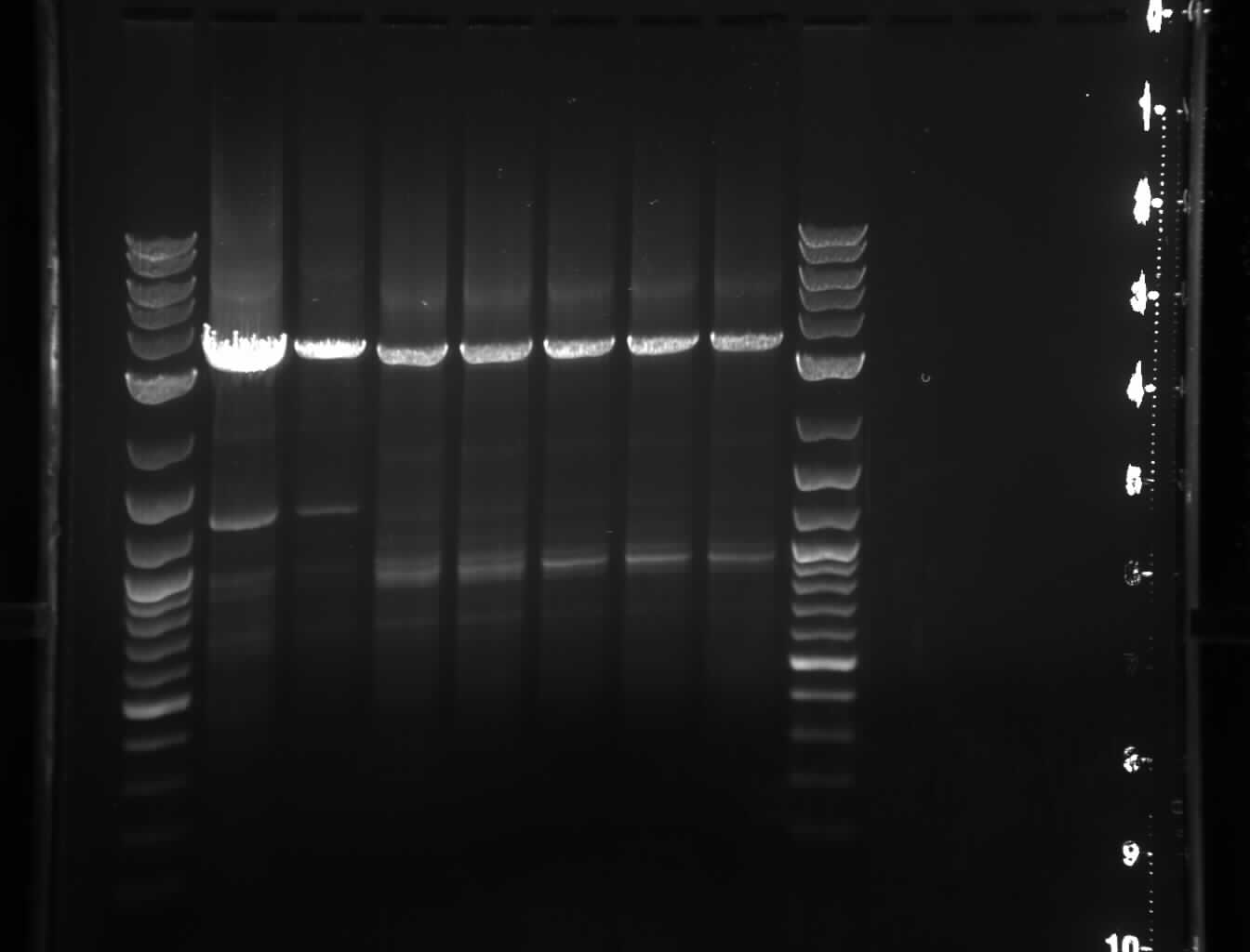{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}