BISC220/S10: Mod 1 Lab 2

Purifying and Handling Proteins in the Lab
Solubilization:
The first step in the purification of a specific intracellular protein is extraction of the protein from the cells. Bacterial cells can be broken and their enzymes extracted (solubilized) by a variety of techniques that may involve mechanical techniques such as grinding or chemical lysis of the cells. The objective is to release the desired enzyme from the cells by as gentle a method as possible in order to retain activity of the enzyme.
Isolation and Concentration:
There are several methodologies that can be employed for the purification of enzymes. These include:
- differential solubility
- ion exchange chromatography
- affinity chromatography
- molecular sieve techniques
- density gradients
- electrophoresis
- electrofocusing
Most of these methods rely on differences in charge (sum of positive and negative charge) or molecular weights of the enzymes and other proteins. Generally cruder, less time consuming methods, are used in the initial processing of crude extracts. This is necessary not only because of the large quantities of protein to be processed but also because of the complexity of the protein mixture. As noted above some of these purification procedures are discussed in your text.
A classic method for the partial purification of enzymes from crude extracts is based on the differential solubility of proteins. Proteins remain dissolved in solution because of their charged surface residues (amino acid side chains) which interact with the molecules of the solvent (water). If such interactions are prevented, the protein molecules will interact principally with one another forming huge aggregates that precipitate out of solution. A common method of enzyme precipitation is by the addition of inorganic salts such as ammonium sulfate.
Stabilization During Purification:
When working with enzymes, it is important to provide environmental conditions that will reduce the amount of enzyme denaturation that occurs during purification procedures. Denaturation results in loss of enzymatic activity. The following are some of the conditions that must be controlled:
- pH: Enzymes have multiple charges on their surfaces. These charges must be preserved to maintain the native structure and enzymatic activity. Buffers are used to maintain the pH of enzyme solutions at a desired pH. The buffer must be in the appropriate concentration, have the correct pKa, and must not adversely affect the protein. See your general chemistry text for a review of buffers and other inorganic chemistry terms.
- Ionic Strength : The ionic strength of the buffer solution is usually critical. Many enzymes require a medium that has a greater ionic strength than provided by the buffer alone. Inorganic salts such as potassium chloride or sodium chloride, which increase the ionic strength, are commonly added to increase the stability of enzymes. Adding sucrose or glycerol to the buffer can often stabilize enzymes requiring a more hydrophobic environment. We will be using glycerol to stabilize β-galactosidase during storage.
- Protection Against Sulfhydryl Oxidation: Enzymes may contain many sulfhydryl groups (SH). One or more may be required for the activity of an enzyme. If these sulfhydryl groups become oxidized, they form intra- or intermolecular disulfide bonds. If necessary, the most effective method of retarding such oxidation is the addition of a reducing agent to the buffer. Dithiothreitol, dithioerythritol and β-mercaptoethanol are among the most effective reducing agents used. β-galactosidase seems to be most stable in a reducing environment, so often β-mercaptoethanol is included in the solutions used to extract and assay the enzyme.
- Protection Against Heavy Metals: In addition to oxidation, sulfhydryl groups may react with heavy metal ions such as lead, iron or copper. Principal sources of metal ions are the reagents used to make up buffers, substrates, and water itself. Deionized distilled water is used to make up reagents and a chelating agent such as EDTA (ethylenediaminetetraacetic acid) can be added as well.
- Temperature: A rule of thumb is to keep enzymes as close to 0-4°C as possible during purification procedures. A few enzymes exist, however, that denature or inactivate at cold temperatures.
Basic Rules for Handling Enzymes
The following information is taken from a popular primer, now published by Roche Pharmaceuticals, but first published in the December 1985 issue of BMBiochemica. by Boehringer Mannheim Biochemicals.
- For best stability, enzymes should be stored in their original commercial form (lyophilized, ammonium sulfate suspension, etc.), undiluted and at the appropriate temperature specified on the label.
- For enzyme solutions and assay buffers, use the highest purity water available. Glass distilled water is best. Deionized water, especially if passed through an old filter or a reverse osmosis device, may contain traces of organic contaminants which inhibit enzymes.
- Enzymes should be handled in the cold (0-4°C) Dilute for use with ice-cold buffer or distilled water as appropriate for each enzyme. While using an enzyme solution or suspension at the bench, keep it in on ice.
- Dilute enzyme solutions are generally unstable. The amount of enzyme required for the experiment should be diluted within 1-2 hours of use. Enzymes should not be diluted for long-term storage.
- Enzymes, especially those that have been diluted, should be checked for activity periodically to ensure that any slight loss in activity is taken into account when designing an experimental protocol. Expiration dates on vials only refer to enzymes stored in the original form at the correct temperature.
- Do not shake crystalline suspensions (e.g. ammonium sulfate suspensions) since oxygen tends to denature the enzyme. The material should be resuspended with gentle swirling or by rolling the bottle on the lab bench. Once the enzyme crystals have been uniformly resuspended, remove the amount needed with a pipette. In many cases, the enzyme crystals may be used directly in the assay procedure.
- Do not freeze crystalline suspensions. Freezing and thawing in the presence of high salt concentrations causes denaturation and loss of activity.
- Vials containing lyophilized enzymes (as well as cofactors, such as NADH and HADPH) should be warmed to room temperature before opening. This prevents condensation of moisture onto the powder, which can cause loss of activty or degradation. If the reagent is hygroscopic, one such mishandling may well ruin the entire vial.
- Avoid repeated freeze-thawing of dilute enzymes and lyophilizates in solution. Store in small aliquots. Thaw one portion at a time and store that portion once thawed at 4C. The stability of individual enzymes may vary greatly and often should be determined empirically under your exact conditions.
- Detergents and preservatives should be used with caution, since they may affect enzyme activity. Sodium azide, for example, inhibits many enzymes which contain heme groups (e.g. peroxidase). Detergents added at concentrations above their critical micellar concentration form micelles which may entrap and denature the enzyme.
- Enzymes should be handled carefully. To avoid contamination of any kind, use a fresh pipette for each aliquot that is removed from the parent vial. Never return unused material to the parent vial. Wear gloves to prevent contaminating the enzyme with proteases, DNAses, RNAses, and inhibitors often found on fingertips. Never pipette by mouth.
- Adjust the pH of the enzyme buffer at the temperature at which it will be used. Many common buffers (Tris, glycylglycine, Bes, Aces, Tes, Bicine, Hepes) change rapidly as the temperature changes. For instance, Tris buffer decreases 0.3 units of pH for EVERY 10°C rise in temperature. A solution of Tris, adjusted to pH7.5 at +25°C will have a pH of 8.1 at 4°C or 7.2 at 37°C. The change in pH per 10°C temperature change for other buffers is: Aces, -0.20; Bes, -0.16 ; Bicine,-0.18; glyclyglycine, -0.28; Hepes,-0.14; Tes, -0.20 [Good, NE et al, (1966) Biochemistry 5:4-7.
- The absorbance at 280nm, widely used to quickly determine the protein concentration of an enzyme solution, actually is due to the presence of tyrosine and tryptophan in the protein. If an enzyme (e.g. superoxide dismutase) contains low amounts of these two amino acids it will not absorb significantly at 280nm.
Detailed information is available on many enzymes. The following are excellent resources.
Resources:
Methods in Enzymology, published by Academic Press, Editors in chief: Sidney P Colowick and Nathan O. Kaplan. There are more than 165 volumes in this series, covering an extensive range of topics.
The Enzymes, 3rd edition, edited by Paul D. Boyer. An excellent, broad series which focuses more on the properties of the enzyme and less on methodology than Methods in Enzymology.
Methods in Enzymatic Analysis, 3rd edition. Editor-in-chief: Hans U. Bergmeyer, published in Verlag Chemie. In-depth discussions of techniques of analysis that use enzymes or that assay enzymatic activity.
Affinity Chromatography and the Determination of the Specific Activity of β-Galactosidase
Affinity Chromatography
The use of metal chelate affinity chromatography for protein purification was first reported by Porath and colleagues in 1975. This landmark report applied the knowledge that histidine and cysteine form rather stable complexes with some cations such as zinc and copper ions. We now know that the amino acid tryptophan shares this characteristic with histidine and cysteine and that some proteins have specific binding sites for these metals. Porath et al (1975) devised a method to tightly bind metal ions to a solid matrix. Agarose beads can be used as such a matrix in a column through which protein solutions are passed. Some proteins in these solutions bind to the matrix but can be specifically eluted by a low pH wash solution. Later work (Porath and Olin, 1983; Kagedal, 1985) showed that it is possible to release proteins from such columns by use of a strong soluble chelator such as EDTA or by chemically competing with the binding. For example, histidine binding can be disrupted by including imidazole in the protein elution buffer. The chemical structure of imidazole is similar to the ring structure of histidine, therefore it competes for the binding site on the cation matrix.
In today’s lab, we are using agarose beads chelated with nickel to fractionate the 6xHis-tagged β-galactosidase. Nickel effectively binds histidine, and the combination of 6 histidine residues in a row at the amino terminus of the β-galactosidase molecule should lead to rather tight binding. After washing the nickel chelated agarose to remove any unbound or loosely bound protein, we will use a buffer containing 200 mM imidazole to release the 6xHis tagged protein from the nickel chelated agarose beads.
Specific Activity
The purification of an enzyme is an attempt to enrich the extract for the desired enzyme while eliminating other cellular components, notably other proteins. One measure of the success of a purification step can be obtained by assaying the activity of the desired enzyme at saturating substrate concentration relative to the total amount of protein present. Specific Activity of an enzyme (also sometimes referred to as maximum velocity, Vmax) is defined as the amount of product formed/unit time (enzyme activity) per milligram (mg) of protein. An enzyme’s specific activity can be employed to evaluate the relative purity of fractions obtained during the purification. In order to evaluate the success of your purification of β-galactosidase, you must measure both the amount of β-galactosidase activity and the total amount of protein (mg) present in the starting material and in your final purified product.
Specific Activity = amount of product formed/unit time/mg protein
β-galactosidase specific activity is often expressed as µmoles of product (ONP) formed per minute per mg of protein.
Specific Activity of β-galactosidase = µmolesONP/minute/mg protein
Once you know the specific activity of your crude extract and your purified fraction, you can proceed to calculate other values useful in determining the success of a purification step such as:
- total activity = (specific activity) x (total mg protein in preparation)
- % yield – the amount of protein of interest retained in the purified fraction
= (total activity of the purified fraction/total activity of the starting material (crude extract)) * 100
3. purification factor – the fold increase of protein of interest in the purified fraction compared to the crude extract
= (specific activity of the purified fraction/specific activity of the starting material (crude extract))
Table I shows the results obtained during a β-galactosidase purification by researchers, Wallenfels et al. (1959), working with β-galactosidase. The inverse relationship between total activity and specific activity is clear. The yield (12%) was reasonable and the enzyme was purified substantially (13.5 fold).

References:
Kagedal L (1998) “Immobilized Metal Ion Affinity Chromatography” In Protein Purification 2nd ed. (Janson J-C and Ryden L eds) Wiley-Liss, New York.
Porath J, Carlsson J, Olsson I, Belfrage G (1975) Metal chelate affinity chromatography, a new approach to protein fractionation. Nature 258: 598-599.
Porath J, Olin B (1983) Immobilized metal ion affinity adsoprtion and immobilized metal ion affinity chromatography of biomaterials. Serum protein affinities for gel-immobilized iron and nickel ions. Biochemistry 22: 1621-1630.
Wallenfels K, Zarnitz ML Laule G, Bender H, Keser M (1959) Biochem Z 331: 459.
To Do Today
In this laboratory session you will purify β-galactosidase from the cell pellet of the genetically modified E. coli that you induced last week to over express this enzyme. You will then determine the total protein content using a modified Bradford dye assay (Bio-rad assay) and assess β-galactosidase specific activity of both the crude extract and the affinity purified fraction using a functional enzyme activity assay. The β-galactosidase specific activity assay uses saturating substrate concentrations of an artificial substrate (ONPG) of β-galactosidase in order to determine, directly, the amount of product (ONP) formed, and more importantly, but indirectly, the concentration of our protein of interest: β-galactosidase. We cannot measure β-galactosidase in mg/ml as easily we can the total protein content. We can, however, use the direct proportionality of absorbance to concentration to calculate product formation/unit of time during a β-galactosidase catalyzed reaction. The functional units, moles of product formed/min reaction time/mg of total protein, are directly proportional to concentration of the enzyme IF the reaction is carried out at saturating substrate concentrations and IF the absorbance of the product is within the accurate range of the spectrophotometer. For the Hitachi spectrophotometers, that range is 0.1-1 Absorbance units. Hitachi Spec Instructions. You will test several different dilutions of both your starting material and your purified fraction in order to find a dilution of enzyme in each that will yield absorbance units within the appropriate range during a 5 minute reaction. There is an equation and sample calculation Enzyme Specific Activity or Velocity to help you convert the units of measurement, absorbance at 420nm to units of specific activity, micromoles of ONP/min/mg protein. As you can see, the total protein concentration determined from the modified Bradford dye assay (Bio-Rad) for total protein is used in the formula for β-galactosidase specific activity.
Specific activity is, therefore, a way of comparing the concentration of the protein of interest to the total protein concentration. When specific activity of the purified product (PF) is divided by the specific activity of the starting material (CE) an evaluation of how much the purification was able to concentrate the enzyme and remove contaminates is obtained (purification factor). Total activity is specific activity multiplied times the amount of protein, therefore, TA comparison of CE and PF help evaluate how much β-galactosidase you retained (% yield). You should create a table with two rows, set up like Table 1, for your data as part of your homework assignment. This table is a succinct analysis of the success of your induction and your one-step purification by affinity chromatography.
Protocol for the Purification of 6xHis Tagged β-galactosidase Affinity Column Purification Protocol
Using B-Per 6xHis Fusion Protein Purification Kit©, product #78100 by Pierce, Inc.
Microsoft Word File: Media:Purification of 6xHis Tagged β gal Protocol.doc
- Defrost the frozen pellet of E.coli BL21(pET-14b) cells prepared in the previous lab session and add 10.0 ml of B-Per Protein Extraction Reagent™, a mild anionic detergent (the exact ingredients of which are proprietary), to the centrifuge bottle. Add 20μl of DNAase (to achieve a concentration of 1 unit/ml from the 5000units/ml stock) to the B-Per and cells. Resuspend the cells by vortex mixing and pipetting up and down until the pellet is off the side of the bottle and all clumps are gone. Try to avoid bubbles. This should take about 5 minutes total, of alternately mixing and letting the mixture sit in your ice bucket. Your goal is to dissolve the cell walls and membranes which will lyse the cells and allow degradation of the DNA. Ask your instructor to check your suspension before proceeding to step 2. If you have obvious clumps, the cells are less likely to be lysed sufficiently.
- Shake the suspension gently for 10 minutes at room temperature using a platform shaker. During this time the lysed cells will release their contents and soluble bacterial proteins will be dissolved in the lysate. Be sure to break up any obvious clumps with gentle vortexing.
- Carefully pour the entire suspension into a Corex tube fitted with a rubber adapter and centrifuge for 15 minutes at 10,500 rpm in the Sorval refrigerated centrifuge at 4C using an SS-34 rotor. You will need to make a balance tube with water. Be sure and record in your lab notebook the speed as g force (use the chart on the wall near the centrifuges to make the conversion).
- Pour the supernatant only into a 15 ml graduated conical centrifuge tube. This solution of dissolved proteins in B-Per reagent is your cell free or Crude Extract. The pellet is bacterial cell debris that can be dumped into your waste container. The Corex tubes are NOT disposable. Please rinse them out and leave them on your bench or return them to your instructor.
- Determine and record the total volume of the crude extract (CE) using the volume markings on the tube. Pipet 600 µl of the CE into a microfuge tube labeled with your team color, lab day, initials and "Crude Extract". Store the aliquot on ice. At the end of lab today, you will give what remains of this aliquot to your instructor to freeze for use next time.
- Add 1000µl (1ml) of the Nickel-Chelated Agarose to the volume of Crude Extract remaining in the 15ml conical tube. It is essential that the agarose suspension be properly mixed before removing an aliquot, so mix it well by swirling or gentle vortexing just before removing it from the stock.
- Shake the Agarose-CE mixture gently for 10 minutes at room temperature on the platform shaker. Make sure the cap is secured tightly. Centrifuge for 5 minutes at top speed in a clinical centrifuge.
- Using a Pasteur pipette, carefully remove and discard the supernatant solution into a waste container provided. Do not try to pour off the supernatant! The 6xHis tagged proteins should now be bound to the nickel- chelated agarose beads at the bottom of the tube.
- Add 3.0 ml of Wash Buffer #1® to the agarose beads in the conical tube and resuspend using a pipette. Do not vortex. Gently shake the suspension at room temperature for 5 minutes on the platform shaker. Centrifuge for 3 minutes at 2500rpm. Carefully remove supernatant and discard.
- Add 3.0 ml of Wash Buffer #2® to the centrifuge tube and resuspend the agarose using a pipette. Do not vortex. Gently shake the suspension at room temperature for 5 minutes on the platform shaker. Centrifuge for 3 minutes. Carefully remove supernatant and discard.
- Add 250µl of Wash Buffer #2® to the agarose, resuspend completely and transfer all of this suspension from the centrifuge tube to a B-PER™ Spin Column that has been placed into one of the special collection tubes provided. To insure that the agarose beads don’t get stuck in the pipet tip of your P1000, cut about a 1/4 inch off the end of it with a single edged razor blade. Centrifuge for 2 minutes at maximum speed in a microcentrifuge. Discard the collection tube with the flow-through.
- Place the spin column in a new collection tube and add 500µl of Elution Buffer containing imidazole. Mix the agarose and the buffer in the spin column using the NOT DISPOSABLE plastic mixer provided by your instructor. Incubate the spin column at room temperature for 5 minutes. During this time, most of the 6x His tagged protein will be eluted from the nickel-chelated agarose into the buffer. After the 5 minute incubation period, recover the protein by centrifuging for 2 minutes at top speed in a microcentrifuge. Your purified fraction will be the flow-through in the collection tube. If you have considerably less than 0.5ml, respin the column to collect more volume.
- Take the spin column out of the collection tube and discard the spin column. Do not discard the material in the collection tube. Using a Pasteur pipet, transfer all of the contents of this collection tube containing the purified beta-galactosidase to a new microfuge tube with volume markings. Record the volume in your notebook! Label the tube to indicate that this is the purified β-galactosidase fraction ("PF β-gal") and save it in your ice bucket.
DO NOT DISCARD ANY OF THE CRUDE EXTRACT OR PURIFIED FRACTION!
You are now ready to assay both the Crude Extract and Purified Fraction for total protein, using a modified Bradford Dye Assay (Bio-Rad assay). You will also assay both for β-galactosidase activity using a different assay that measures ONP production.
Modified Bradford Dye Assay (Bio-Rad™ Assay) for Total Protein
Microsoft Word File: Media:Modified Bradford Dye Assay Protocol.doc
Testing the quality of the purification:
The total protein assay of choice is a spectrophotometric method known as the modified Bradford dye assay. It is also sometimes called the “Bio-Rad™ Assay" because the assay reagent was purchased from a company named Bio-Rad. It is a dye-binding assay based on a differential color change of Coomassie Brilliant Blue in response to various concentrations of protein. The absorbance maximum for an acidic solution of Coomassie Brilliant Blue G-250 shifts from 465 nm to 595 nm when binding to protein occurs. In order to determine protein concentration in unknowns (CE and PF) we must compare their absorbances to the absorbance of known concentrations of protein that have been used to create a standard curve. Please note that this assay does not measure only our protein of interest, β-galactosidase, but instead, measures the concentration of all the proteins, including β-galactosidase, that are present in our starting and our purified material. We measure total protein for two reasons: we need to use the total protein concentration determined by this assay in order to determine the concentration of β-galactosidase in our specific activity assay and because knowing the total protein concentration in our starting vs. purified material helps us determine how much contaminating protein we removed during our purification.
Dilution Preparation for the Standard Curve
Prepare a set of 6 microfuge tubes. Dilute a stock solution of bovine serum albumin (BSA) with a concentration of 1mg/ml to yield 200 microliters of BSA in concentrations of: 0.1, 0.2, 0.4, 0.6, 0.8 and 1 mg/ml.
Table 2: Dilution of Stock BSA to make protein standards for total protein assay
| Tube # | BSA Concentration | Z Buffer in μL | Stock BSA |
|---|---|---|---|
| 1 | |||
| 2 | |||
| 3 | |||
| 4 | |||
| 5 | |||
| 6 |
The diluent will be Z-buffer (60mM Na2HPO4, 60mM NaH2PO4, 1mM MgSO4, 0.27% beta-mercaptoethanol) and the final volume needed is 200 μl. The formula C1 x V1 = C2 x V2 should help you. Check with your instructor to confirm your dilution strategy in the table above before making these working concentrations of the standards. Remember to add the smaller volume to the larger volume.
Dilution Preparation of Unknowns:
Dilute your CE and PF 1:5 in Z buffer to yield a final volume of 300 μl of each.
We are trying to determine the concentration of protein in each but since we don’t know it at this point we will have to use a different dilution formula.
sV x df = ftV where sV is starting volume (the unknown); df is the dilution factor (5); ftV is the final total Volume (300μl). Check your dilution strategy with your instructor before making the dilutions.
Amount of Z-buffer required = ?
Amount of CE or PF required = ?
Total Protein Assay Protocol:
- Label a set of 12 glass tubes. See Table 3 below for the contents of each tube. Put the Protein Assay Reagent into all tubes last. Cover the tubes with parafilm and mix well by vortexing.
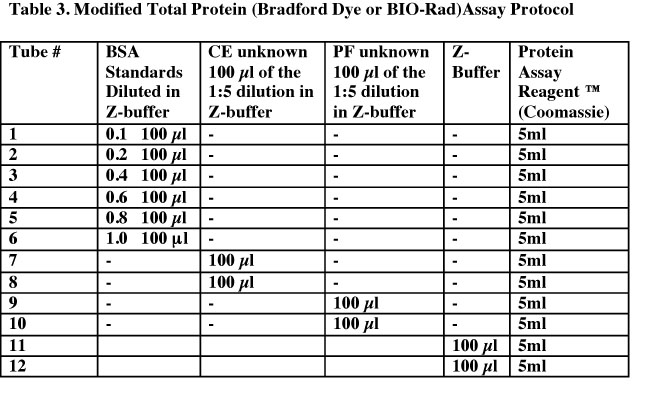
- Let the reactions and blanks incubate at RT for a minimum of 15 minutes. The reaction is stable for one hour.
- You can start making your dilutions or set up your assay tubes for the specific activity assay while you are waiting for the total protein assay to maximize its absorbance shift.
- After maximum color development (at least 15 minutes but not more than one hour) mix all 12 tubes again and transfer enough of the contents of each assay tube or blanks to a set of labeled 1.5mm plastic cuvettes so that the cuvettes are at least 2/3 to 3/4 full. Check to be sure that there is no precipitate interfering with light passage. If so, let the precipitate settle to the bottom before reading absorbance.
- Follow the directions in the appendix for reading absorbance using either the Hitachi or Cary 50 spectrophotometers. Set the wavelength to 595nm.
- Blank the spectrophotometer using the reagent blank(s) (tubes 10, and or11).
- Read A595 for all tubes and record in your lab notebook.

Tubes 1-6 (the diluted BSA standards) contain known concentrations of protein. You will use those known concentrations and absorbances to create a standard curve and linear regression line using Excel (see Linear Regression Instructions). The creation of a standard curve will allow calculation of the protein concentration in mg/ml from the absorbance of the diluted CE and PF. Make sure your R2 value for the line is at least .95. The CE and PF have been assayed in duplicate, therefore, the concentrations can be averaged if the replicates are within 10% of each other. Record the total protein concentration for both the CE and the PF in your lab notebook. Remember that the true concentration will be 5 times the value obtained from your linear regression formula since the assay used a 1:5 dilution.
Make sure that the 1:5 dilution was appropriate before proceeding to the specific activity assay. If it was, the absorbance readings will be within the range of concentration of the standards used to create the standard curve. If the standards were diluted properly, they will give an absorbance reading within the accurate range of the spectrophotometer (0.1-1 A for the Hitachi specs). If your absorbance readings are outside this range, you will have to repeat the assay using different dilutions in order to obtain an accurate total protein concentration for your CE and PF. The protein concentration of the PF and CE will be used to convert absorbance to specific activity units in the following assay for determining how much beta-galactosidase there is the purified fraction and in the starting material from which it was made (CE). Knowing total protein concentration and the specific activity of beta-galactosidase in the CE and PF will allow us to determine the success of our affinity chromatography purification.
DO NOT DISCARD ANY OF THE CRUDE EXTRACT OR PURIFIED FRACTION!
Determining the amount of β-Galactosidase in the Total Protein by β-Galactosidase Enzyme Activity Using A420 measurement of ONP production
Microsoft Word File: Media:Determining the amount of β gal by enzyme activity.doc
In-vivo, β-galactosidase cleaves lactose to yield galactose and glucose, but in vitro, the appearance of the products of galactose and glucose are difficult to monitor. The colorless compound ortho-nitro-phenyl-galactoside (ONPG) is substituted for lactose and yields upon hydrolysis (cleavage) by β-galactosidase a yellow compound, ortho-nitrophenol, (ONP) and galactose. The addition of concentrated sodium carbonate (Na2CO3) shifts the pH to a very basic pH 11, a condition which inactivates the enzyme. The amount of colored product (ONP), formed from the colorless substrate (ONPG), can be quantified using a spectrophotometer and then converted to a concentration of ONP using its molar extinction coefficient ( Enzyme Specific Activity or Velocity for a sample calculation).
The total protein assay you performed has allowed you to determined the protein concentration of your two fractions (the crude extract and the purified fraction) but you do not know how much of that protein is our enzyme of interest, beta-galactosidase. You will now perform a specific β-galactosidase activity assay on a series of dilutions of both the crude extract (CE) and of the purified fraction (PF) in order to determine the concentration of enzyme that will yield an absorbance in the most accurate range of measurement of the spectrophotometers (0.1-1.0 for the Hitachi spec). Our goal is to find a diluted form of β-galactosidase in the CE & in the PF that gives an absorbance reading of close to 0.5A in the specific activity assay.
Suggestions for appropriate dilutions based on previous experimentation are: CE: 1:50; 1:100; 1:200; 1:400. Since there is more concentration beta-galactosidase in the PF, dilutions of: 1:100, 1:200; 1:400; and 1:800 should be tested.
Making a serial dilution of the CE and PF for the SA assay:
Since you have a limited volume of both crude extract and purified fractions and you will need a minimum of 100 µl for each assay, it is advisable to make a little more than you need of each dilution but not so much more that you waste your fractions. It is perfectly acceptable, and often preferable, to use part of a stronger concentration to make the next weaker one.
For example, let's assume you want to end up with 250 µl of each dilution. You could start by making a 1:50 dilution from the 1:5 dilution of the CE or PF you prepared for the Bradford dye assay. Making a 1:10 dilution of some of this previously made 1:5 dilution will give you the 1:50 you desire. How could you make total volume of 500µl of a 1:50 dilution? Use the formula:
Sv (starting volume, the unknown) x dilution factor (in this case,10) = tV (total volume, 500µl).
After preparing 500µl of a 1:50 dilution and mixing well you could use 250µl of that 1:50 dilution added to an equal volume of buffer to make 500µl of a 1:100 dilution. If you wanted to continue to dilute with buffer equal volumes of each sequentially weaker concentration, you would end up with 250µl of each of the concentrations desired: 1:50, 1:100, 1:200, 1:400, 1:800. This is a serial dilution.
Please show your dilution strategy to your instructor before you proceed.
After your instructor has approved your dilution strategy and you have on ice all of your labeled microfuge tubes with each of the specified dilutions of the CE and PF in Z-buffer (60mM Na2HPO4, 60mM NaH2PO4, 1mM MgSO4, 0.27% beta-mercaptoethanol), you are now ready to start the assay.
Protocol for Assay of Specific Activity (at saturating substrate concentration)
- Label a new set of 8 glass tubes with the 4 dilutions of CE and the 4 dilutions of PF to be tested. For the CE, you will test: 1:50, 1:100, 1:200, 1:400. For the PF, test: 1:100. 1:200, 1:400, 1:800. Label two additional tubes #10 B (reagent blanks for the spectrophotometer).
- Pipet 1.9ml of Z-buffer into the 8 glass test tubes prepared above. Pipet 2ml Z-buffer into the 2 reagent blanks (#10B).
- Pipet 100μl of the appropriate enzyme dilution into the labeled tube containing Z-buffer prepared in #1. Mix well by vortexing. Put NO enzyme in the blanks!
- Equilibrate all 10 tubes to 28C in a water bath. Five minutes should be sufficient.
- Start the reaction by adding 400 µl (0.4 ml) of substrate (ONPG, 4mg/ml) to the first tube, vortex immediately, and quickly return the tube to the water bath. In order to insure that all reactions occur for exactly the same amount of time, add 400µl (0.4 ml) substrate at carefully timed intervals, such as every 20 or 30 seconds. What is the effective concentration of ONPG?
- At exactly 5 minutes after adding ONPG to the first tube, start adding 1000µl (1 ml) of stop buffer, 1M Na2CO3 to each tube in the same order at the same time interval. What is the effective concentration of stop buffer? Mix well after each addition. Since you will be calculating specific activity of beta-galactosidase as µmoles of product formed (ONP)/ minute/mg of total protein, timing of the reaction is critical.
- Pour some of each tube into a set of labeled cuvettes. Make sure the cuvettes are 2/3 to ¾ full. It doesn’t matter if each has exactly the same volume.
- Read A420 in the spectrophotometer. Don’t forget to change the wavelength from 595nm to 420nm. Zero the instrument using the reagent blanks.
- Calculate specific activity from absorbance using the Beer-Lambert formula. The molar extinction co-efficient of ONP is 4800 M-1 cm-1 and the path length of the cuvette used is 1 cm. The concentrations and total protein content in each of your fractions were determined by the Bradford dye assay. There is a sample calculation Enzyme Specific Activity or Velocity.
Sample Storage
- Transfer 50 µl from the microfuge tube of undiluted purified fraction (PF β-gal) into a new microfuge tube labeled with your team color, initials, lab day, and clear indication of what is in the tube. This aliquot will be saved for SDS-PAGE in Lab IV.
- Determine the remaining volume of the purified fraction and add an amount of 70% glycerol equal to 1/2 the volume of that fraction. Mix well using the vortex. For example, if your purified fraction has a volume of 250 µl, you would add 125 µl of 70% glycerol solution it. The glycerol acts to preserve enzymatic activity while the samples are frozen. We will be using this aliquot of purified fraction to study the enzyme kinetics of β-galactosidase in Lab III.
- Give the 2 aliquots (diluted in glycerol and 50µl undiluted) of PF prepared above to your instructor. Also give your instructor your remaining Crude Extract. All 3 samples will be frozen at -70oC for use later in the series. Make sure all samples are labeled with the contents (differentiate the one with glycerol), your team color, initials and date.
