Todd:Chem3x11 ToddL1

Chem3x11 Lecture 1
This lecture is an overview of isomerism, then some material on stereocentres and what chiral molecules look like in 1H NMR spectroscopy.
(Back to the main teaching page)
Key concepts
- There are two different kinds of stereoisomers
- Conformations of alkanes governed by sterics and electronics
- Centres that are "nearly chiral" become interesting in a chiral environment
Kinds of Isomerism
Constitutional isomers are the easiest to think about. The molecules are put together differently. Interview each atom in Molecule A in turn by asking "What other atoms are you bonded to?", then do the same for each atom in Molecule B. If the answers you get are different, Molecules A and B are constitutional isomers.

Atoms in stereoisomers would give you the same answer since the atoms are connected to each other the same way, only the positions of the atoms in space changes. The easiest kind of stereoisomers to conceptualize are those around a double bond, i.e. E- and Z- isomers. To interconvert these you'd need to break the double bond, and when a bond needs to be broken we call that configurational isomerism.

Stereocentres are like that too, so enantiomers are configurational isomers.

Stereoisomers you can interconvert without breaking any bonds are both interesting and not. The example below concerns alkanes, which seems silly because we never isolate the alkane isomers under normal conditions - the bond rotation is too easy/fast. But there are some very interesting and important examples of this kind of isomerism, particularly in Nature. Isomers of this kind, where all the atoms are connected to each other the same way, but there is a difference in the 3D arrangement of atoms BUT the isomers can be interconverted without breaking any bonds are called conformational isomers.

An Example of Conformational Isomers - The Rotations of Alkanes
Alkanes (acyclic ones) can rotate about each C-C bond. The different arrangements in space can be interconverted without breaking any bonds, and we call these structures conformational isomers. Typically the interconversion is super-fast and we can't isolate the separate isomers. If it becomes possible to see the separate isomers by e.g. 1H NMR spectroscopy we sometimes call them rotamers, or, more formally and generally atropisomers.
Different conformational isomers have different arrangements of atoms in space, and these will have different energies, e.g. ethane, below.

Why do these isomers interconvert so quickly? Pretty low barrier to rotation:

For butane there is a greater preference for the conformation where the methyl groups are far apart. Everything's still moving very quickly, but the extended conformation (where the methyl groups are as far from each other as possible) is the lowest in energy. That's why we draw alkane chains as zig-zags, typically.

When butane is in one of its eclipsed conformations, the steric clashes explain the high energies.

In the staggered conformations, we should just learn a couple of commonly-used terms: gauche and antiperiplanar

The gauche conformation of butane still has a bit of a steric clash, and obviously the clashes are going to be lowest in the antiperiplanar conformation, which becomes our mental picture of butane, as the statistically most likely conformation.

Butane (and lots of other alkanes) are therefore easily understood in terms of sterics. But that's not the complete story. As with everything in organic chemistry, there is always a mixture of sterics and electronics. In ethane, for example, there are no "bulky" groups providing any steric clashes. There is still a small preference for the staggered conformation. But actually the energetic cost of the eclipsed conformation is surprisingly large, given that there are only H's "clashing" here.

To understand this, we should look at the orbitals involved. We humans draw a bond as a thin line. Remember it's a cloud of electrons, so a cloud of negative charge. So bonds don't like to be near each other. This is just another way of talking about a steric clash. But the fact that the H's in ethane are small shouldn't make us forget that the C-H bonds prefer not to be near each other either. So the eclipsed conformation is disfavoured because of a clash of the bonding electrons in the eclipsed bonds. We call this torsional strain.

There's something else, which is more subtle and which becomes important in lots of other cases we don't have time for. There is actually a benefit to the staggered conformation. The electrons in one C-H σ bond can overlap with the empty C-H σ* bond on the adjacent position. This would seem like a problem, but it's not. Any two-electron interaction between a filled orbital and an empty orbital is energetically favourable (i.e. bonding). The specific case when a C-H σ bond overlaps with an adjacent empty orbital is sometimes called hyperconjugation. If you had me as a lecturer in 1102, I told you about this when we learned by tertiary carbocations are more stable than primary. You can read more about the research that suggests hyperconjugation is the key factor in this paper.

A molecule is a complex electronic structure, and unexpected interactions between orbitals in three dimensions can explain a whole range of things that may seem puzzling when molecules are drawn as dull 2D objects.
Some Example Rotational Barriers
It can be useful and interesting to compare barriers to rotations around various single bonds (Scheme 14), to appreciate how many times per second some conformers are interconverting, and the size of the rotational barrier needed to allow the conformers to be distinguishable by e.g. NMR spectroscopy or be actually isolable (at which point we call them atropisomers)
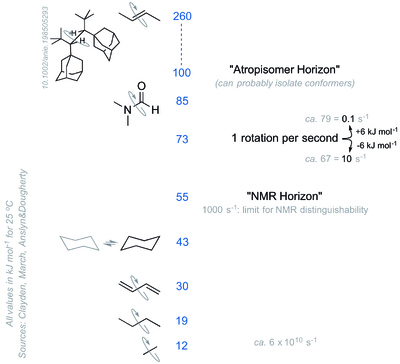
Conformations of Alkanes in NMR Spectroscopy
Fast rotation around single bonds means we don't see separate conformations of alkanes in NMR spectroscopy. If we could stop the movement of the molecules, we would see the different conformations as different.
Configurational isomers are interesting. Enantiomers look the same by NMR spectroscopy (with one condition, which we'll come to). Diastereomers (configurational isomers that aren't mirror images, e.g. double bond isomers) look different by NMR spectroscopy.
But things are a little more interesting than that. To understand when atoms/molecules/centres go from being identical by NMR spectroscopy to becoming distinct, let's examine three situations, of increasing complexity.
Symmetric Centres
This molecule is symmetric:

The two H's can be interconverted just by rotating the molecule 180° around an axis. The H's are always chemically identical. They are called homotopic.
Symmetric Centres that Become Asymmetric in a Chiral Environment
This molecule is symmetric, this time about a plane of symmetry (the plane of the page):

When examined under normal conditions by 1H NMR spectroscopy, the H's look the same as each other - they are equivalent. The H's become distinguishable in a chiral environment. The H's are enantiotopic or heterotopic. Here are two ways to put that centre in a chiral environment:
A) Put it in an Enzyme Active Site
An enzyme active site is chiral. The enzyme can distinguish enantiotopic H's. If you can't see this, imagine holding a model of this molecule, and think how your hands (which are chiral) could tell which H is which, and how to do that you need to use the other two groups as reference points.

There are lots of examples one can choose of this happening in Nature, e.g. the selective phosphorylation of this symmetric diol, glycerol.

There's also a bunch of examples where oxidation/reduction chemistry happens selectively at centres like this mediated by NAD+/NADH.
B) Put it in a Molecule with a Stereocentre
If the molecule of interest already has a stereocentre in it, then effectively our centre with enantiotopic groups is already in a chiral environment. Thus enantiotopic groups are distiguishable in NMR spectroscopy if the molecule is chiral somewhere, no matter what the distance is between the two parts of the molecule when you draw the molecule on paper. So in this molecule H1 and H2 cannot be interconverted by a plane or axis of symmetry. We call them heterotopic, or more commonly diastereotopic. The protons are different by 1H NMR spectroscopy (they are anisochronous in official terminology that is very rarely used). Not only do they actually give rise to different signals, they will also couple with each other.

Note that one centre needs to have enantiotopic H's and there needs to be another stereocentre. In the following molecule we don't have enantiotopic H's. While it's true that the H's can't be interconverted by an axis or plane of symmetry, the C-C bond rotation is so fast that the H's just see an average of the adjacent centre, meaning they are equivalent by NMR spectroscopy.

So as the C-C bond rotates, the H's do superimpose on each other, but they all experience the same electronic/magnetic environment in the molecule, as a weighted average.

In contrast, in the case where we have diastereotopic H's, then though the H's seem to be superimposing on each other as the C-C bond spins, there isn't quite a perfect super-position. The presence of that methyl group means H1 never quite experiences what H2 experiences, even though the C-C bond is spinning very fast.

Another way of checking for the presence of diastereotopic protons is to draw the molecule where one H has become a D, and then draw the molecule where the other proton has become a D. If your drawings are diastereomers, then your original H's were diastereotopic.

There is a last piece of optional jargon, which is the word we use to describe the molecule in Scheme 15 - the one that is "nearly asymmetric". The molecule is termed prochiral. A carbon centre that is of the form Caabc is prochiral (unless one of a, b or c is the enantiomer of another - see if you can see why that matters). Again, imagine replacing the enantiotopic H's sequentially with deuteriums. That would give a pair of enantiomers. Hence the molecule is prochiral.
There's also terminology you ought to know that is used to describe the two enantiotopic H's. Take one of the H's in our original molecule, below. Make it a deuterium. D has a higher priority than H when assigning the stereocentre. So in the below, when we make H1 D, the centre would be assigned S, meaning H1 is pro-S. H2 is, by the same analysis, pro-R.

This "pro" terminology was developed in 1966. It's widespread. It's slightly ambiguous because a molecule can also have prochiral faces. In organic syntheses we often see this with aldehydes. Often we do nucleophilic additions to aldehydes to make new C-C bonds, converting the aldehyde centre to an alcohol. Typically that generates a chiral alcohol. The aldehyde is termed prochiral even though there is no stereogenic centre. Be aware that this term is used in these two different ways, and if you want to be specific, stick to talking of enantiotopic or diastereotopic H's.
Example of a 1H NMR Spectrum of a Molecule with Diastereotopic H's
This now ought to make more sense:

And the nature of the peaks in this 1H NMR spectrum of this sulfoxide (a real example from my lab) ought to tell you something interesting about the stereochemistry of sulfoxides that is not clear from the way the molecule is drawn.

The Licence for This Page
Is CC-BY-3.0 meaning you can use whatever you want, provided you cite me.
